Variační kvantový eigensolver (VQE)
Pro tento modul musíš mít funkční prostředí Python a nainstalované nejnovější verze následujících balíčků:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Jak tyto balíčky nastavit a nainstalovat, najdeš v průvodci Instalace Qiskitu. Aby sis mohl/a spouštět úlohy na skutečných kvantových počítačích, budeš si muset zřídit účet IBM Cloud podle kroků v průvodci Nastavení účtu IBM Cloud.
Tento modul byl otestován a využil přibližně 8 minut QPU času. Jde o odhad – tvoje skutečná spotřeba se může lišit.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Úvod
Od doby, kdy byl na počátku 20. století vyvinut kvantově-mechanický model, vědci chápou, že elektrony neobíhají kolem jádra atomu po pevných drahách, ale existují v oblastech pravděpodobnosti zvaných orbitaly. Tyto orbitaly odpovídají konkrétním, diskrétním energetickým hladinám, které mohou elektrony obsazovat. Elektrony přirozeně sídlí na nejnižších dostupných energetických hladinách – tzv. základním stavu. Pokud však elektron pohltí dostatek energie, může přeskočit na vyšší energetickou hladinu a vstoupit do excitovaného stavu. Tento excitovaný stav je dočasný a elektron se nakonec vrátí na nižší energetickou hladinu, přičemž pohltěnou energii uvolní, často ve formě světla. Tento základní proces absorpce a emise energie je klíčový pro pochopení toho, jak spolu atomy interagují a vytvářejí vazby.
Když se atomy spojí a vytvoří molekuly, jejich atomové orbitaly se kombinují a vznikají molekulové orbitaly. Uspořádání a energetické hladiny elektronů v těchto molekulových orbitalech určují vlastnosti výsledné molekuly a pevnost chemických vazeb. Například při vzniku molekuly vodíku () ze dvou jednotlivých atomů vodíku obsazuje elektron každého atomu atomové orbitaly. Jak se atomy přibližují, tyto atomové orbitaly se překrývají a kombinují, čímž vznikají nové molekulové orbitaly — jeden s nižší energií (vazebný orbital) a jeden s vyšší energií (protivazebný orbital). Oba elektrony, po jednom z každého atomu vodíku, přednostně obsadí vazebný orbital s nižší energií, což vede ke vzniku stabilní kovalentní vazby držící molekulu pohromadě. Energetický rozdíl mezi oddělenými atomy a vzniklou molekulou, zejména energie elektronů v molekulových orbitalech, určuje stabilitu a vlastnosti vazby.
V následujících částech prozkoumáme tento proces tvorby molekuly, zaměříme se na molekulu . Použijeme skutečný kvantový počítač v kombinaci s klasickými optimalizačními technikami, abychom nalezli energii tohoto jednoduchého, ale fundamentálního procesu. Tento experiment poskytne praktickou ukázku toho, jak lze kvantové výpočty využít k řešení problémů v oblasti výpočetní chemie, a přiblíží roli energie elektronů.
VQE – variační kvantový algoritmus pro problémy vlastních hodnot
Aproximační techniky v chemii – variační princip a bázová sada
Erwin Schrödinger nepřispěl ke kvantové mechanice jen zavedením nového elektronového modelu; v zásadě položil základy vlnové mechaniky tím, že odvodil slavnou časově závislou Schrödingerovu rovnici:
Zde je hamiltonián – operátor reprezentující celkovou energii systému – a je vlnová funkce obsahující veškeré informace o kvantovém stavu systému. (Poznámka: je totální časová derivace a vlastní hodnota energie zde není explicitně zahrnuta.)
V mnoha praktických aplikacích – například při určování povolených energetických hladin atomů a molekul – ale místo toho používáme časově nezávislou Schrödingerovu rovnici (rovnici vlastních hodnot energie), která se odvozuje z časově závislého tvaru předpokladem stacionárního stavu. Stacionární stav je kvantový stav, v němž se hustota pravděpodobnosti nalezení částice v daném bodě prostoru časem nemění.
V tomto tvaru představuje vlastní hodnotu energie odpovídající kvantovému stavu . Hamiltonián zahrnuje různé příspěvky k energii: kinetickou energii elektronů a jader, přitažlivé síly mezi elektrony a jádry a odpudivé síly mezi elektrony.
Řešení rovnice vlastních hodnot energie nám umožňuje vypočítat kvantovaná energetická hladiny atomových a molekulárních systémů. U molekul je však přesné řešení obtížné, protože vlnová funkce , která popisuje prostorové rozložení elektronů, je složitá a má vysoký počet dimenzí.
Proto vědci používají aproximační techniky k získání prakticky použitelných a přesných řešení. V tomto textu se zaměříme na dvě klíčové metody:
-
Variační princip
Tato metoda aproximuje vlnovou funkci a upravuje ji tak, aby se co nejvíce přiblížila cílové energii, obvykle energii základního stavu systému. Klíčová myšlenka variačního principu je jednoduchá:
- Pokud odhadneme vlnovou funkci (tzv. „zkušební funkci"), energie z ní vypočítaná bude vždy rovna nebo vyšší než energie základního stavu () systému.
- Úpravou parametrů ve zkušební funkci lze získávat stále lepší aproximaci energie základního stavu.
- Přesnost metody silně závisí na volbě zkušební vlnové funkce . Špatně zvolená zkušební funkce může vést k odhadu energie, který je daleko od přesné hodnoty.
-
Aproximace bázovou sadou
Druhá aproximační metoda přichází ke slovu ve fázi konstrukce vlnové funkce – jde o přístup bázové sady. V kvantové chemii je přesné řešení Schrödingerovy rovnice pro molekuly prakticky nemožné. Místo toho aproximujeme složitou mnohaelektronovou vlnovou funkci tak, že ji sestavíme z jednodušších, předem definovaných matematických funkcí. Bázová sada je v podstatě soubor těchto známých matematických funkcí, obvykle soustředěných na atomy v molekule, které slouží jako stavební kameny pro reprezentaci tvaru a chování elektronů v systému. Představ si to jako snahu vytvořit detailní sochu pouze ze standardních LEGO kostek – čím více typů a velikostí kostek máš (čím větší bázová sada), tím přesněji dokážeš aproximovat původní tvar.
Tyto bázové funkce jsou často inspirovány analytickými řešeními jednoduchých systémů, jako je atom vodíku, a mají tvar gaussovských nebo Slaterových funkcí, i když jde stále o aproximace. Místo práce s teoreticky „přesnými", ale nepraktickými plnými molekulovými orbitaly je vyjadřujeme jako lineární kombinaci (součet s koeficienty) těchto bázových funkcí. Tato metoda je při použití bázových funkcí připomínajících atomové orbitaly známá jako přístup lineární kombinace atomových orbitalů (LCAO). Optimalizací koeficientů v této lineární kombinaci lze nalézt nejlepší možnou aproximovanou vlnovou funkci a energii v rámci omezení zvolené bázové sady.
- Čím více funkcí bázová sada obsahuje, tím lepší je aproximace, ale za cenu vyšší výpočetní náročnosti.
- Malá bázová sada poskytuje přibližný odhad, zatímco velká bázová sada dává přesnější výsledky za cenu vyšší výpočetní náročnosti.
Shrnutí: abychom zajistili proveditelnost výpočtů a snížili jejich výpočetní náklady, používáme variační princip k aproximaci vlnové funkce, který snižuje výpočetní složitost a umožňuje iterativní optimalizaci minimalizující energii. Přístup bázové sady zároveň zjednodušuje výpočty tím, že reprezentuje atomové orbitaly jako kombinaci předdefinovaných funkcí, místo přímého řešení pro spojitou vlnovou funkci.
Otestuj si porozumění
Uvažuj zkušební vlnovou funkci , kde je normalizační konstanta a je nastavitelný parametr.
(a) Normalizuj zkušební vlnovou funkci tak, že určíš splňující podmínku .
Answer
Normalizace dané zkušební vlnové funkce:
Použijeme gaussovský integrál:
dosadíme a dostaneme:
(b) Vypočítej střední hodnotu hamiltoniánu daného předpisem kde , což odpovídá potenciálu jednoduchého harmonického oscilátoru.
Answer
Hamiltonián harmonického oscilátoru je:
Střední hodnota kinetické energie
Vypočítáme druhou derivaci:
Tedy:
S využitím standardních výsledků gaussovských integrálů:
Střední hodnota potenciální energie
S využitím:
dostaneme:
Střední hodnota celkové energie
(c) Použij variační princip k nalezení optimálního minimalizací .
Answer
Optimalizace pro minimální energii
Derivujeme
Řešíme:
Dosadíme do :
což odpovídá přesné energii základního stavu kvantového harmonického oscilátoru.
VQE (Variational Quantum Eigensolver)
Variační kvantový eigensolver (VQE) je hlavní metoda, kterou budeme používat ke zkoumání procesu . Zde se podíváme na to, co VQE je a jak funguje. Nejdříve ale udělejme krátkou zastávku a zamysleme se nad jednou velmi důležitou věcí prostřednictvím kontrolní otázky.
Ověř si porozumění
Když už máme tolik strategií pro chemické problémy, proč vůbec potřebujeme kvantový počítač? A k čemu slouží kombinace kvantových a klasických počítačů?
Answer
Kvantové počítače mají šanci revolucionizovat chemii tím, že zvládnou problémy, se kterými si klasické počítače nevědí rady kvůli exponenciálnímu škálování kvantových stavů. Richard Feynman proslule poznamenal, že pro simulaci přírody musí být i výpočty kvantové [ref 1].
Například simulace kofeinu s nejjednodušší bázovou sadou (STO-3G) by vyžadovala bitů — to je mnohem více, než je celkový počet hvězd v pozorovatelném vesmíru () [ref 2]. Kvantový počítač dokáže popsat elektronové orbitaly kofeinu pomocí 160 qubitů.
Kvantové počítače přirozeně zpracovávají kvantové interakce pomocí superpozice a provázanosti, což představuje slibný způsob, jak umožnit přesné molekulární simulace. Navíc můžeme kombinovat výhody kvantových počítačů (simulace elektronů) a klasických počítačů (předzpracování a postprocesing dat, správa algoritmického procesu, optimalizace apod.). Od těchto přístupů se očekává, že urychlí objevování materiálů, návrh léčiv a předpovědi reakcí, čímž se sníží nákladné pokusy metodou pokus-omyl. [ref 3][ref 4]
Pokud chceš vědět více o tom, proč jsou kvantové počítače potřebné pro chemické problémy a proč kombinovat kvantové a klasické výpočetní zdroje, podívej se na tyto články:
Nyní se vraťme k VQE.
VQE kombinuje sílu kvantových počítačů s klasickými počítači a k nalezení energie základního stavu systému zásadně využívá variační principy. Abys VQE lépe pochopil(a), rozlož jej do tří částí:
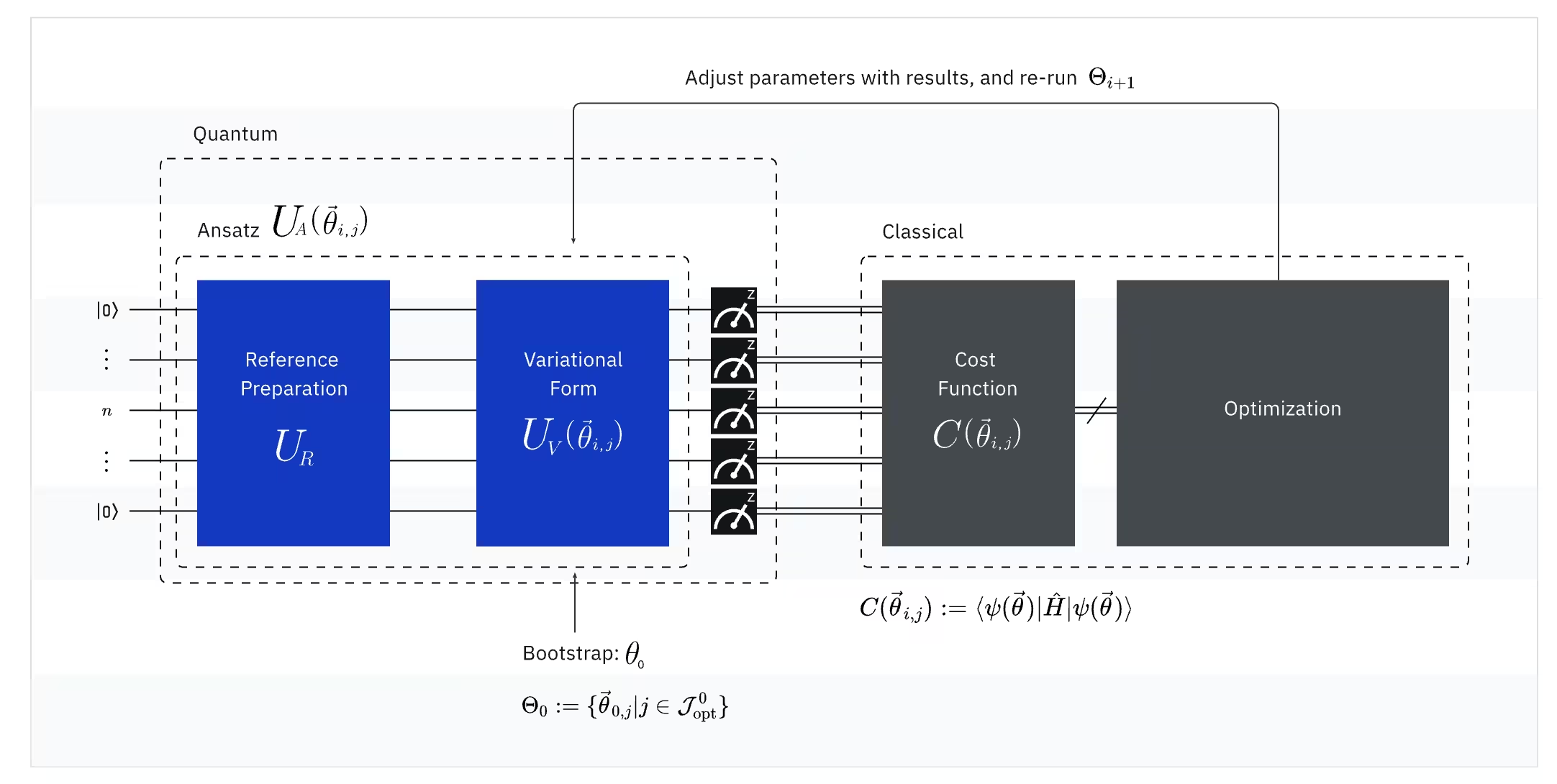
(Kvantová) Observabla: molekulární hamiltonián (energie molekuly)
Ve VQE je molekulární/atomový hamiltonián observablou, což znamená, že jeho hodnotu lze změřit experimentem. Naším cílem je najít nejnižší možnou energii (energii základního stavu) molekuly. K tomu používáme zkušební kvantový stav generovaný parametrizovaným kvantovým obvodem (ansatz). Observablu měříme a kvantový stav optimalizujeme, dokud nedosáhneme nejnižší možné energie.
Bázová sada použitá pro molekulární hamiltonián určuje počet potřebných qubitů a přímo ovlivňuje přesnost VQE. Volba správné bázové sady je klíčová pro vyvážení efektivity a přesnosti. Abychom zjednodušili výpočty bez změny bázové sady, můžeme použít strategie jako uplatnění symetrie a redukce aktivního prostoru. Mnoho molekul má symetrické tvary (jako motýl nebo sněhová vločka), což znamená, že se některé části chovají stejně. Místo toho, abychom vše počítali zvlášť, se zaměřujeme pouze na jedinečné části, čímž šetříme kvantové zdroje — to je využití symetrie. Při redukci aktivního prostoru uvažujeme pouze důležité orbitaly, protože ne všichni elektroni výrazně ovlivňují energii molekuly. Elektrony blízko jádra zůstávají většinou nezměněny, zatímco jiné ovlivňují vazby. Aplikací těchto metod lze VQE zefektivnit při zachování přesnosti.
Jakmile pomocí vhodné bázové sady a výše uvedených strategií získáme molekulární hamiltonián, musíme jej transformovat do podoby vhodné pro kvantové počítače. Mapování problémů na Pauliho operátory může být poměrně složité. To platí zejména v kvantové chemii, která pracuje s nerozlišitelnými částicemi (elektrony), zatímco qubity jsou rozlišitelné. Podrobnostem mapování se zde věnovat nebudeme, ale odkazujeme tě na následující zdroje. Obecnou diskusi o mapování problému na kvantové operátory najdeš v kurzu Quantum computing in practice. Podrobnější diskusi o mapování chemických problémů na kvantové operátory najdeš v kurzu Quantum chemistry with VQE.
Pro tento modul ti poskytneme příslušné (jednoqubitové) hamiltoniány pro a , abychom se mohli soustředit na použití kvantového počítače. Tyto jednoqubitové hamiltoniány jsou připraveny pomocí bázové sady STO-6G a Jordanova-Wignerova mapování, což je nejpřímočařejší mapování s nejjednodušší fyzikální interpretací, protože mapuje obsazenost jednoho spin-orbitalu na obsazenost jednoho qubitu. Také jsme použili techniku redukce qubitů pomocí symetrie hamiltoniánu, která využívá vzorce v chování spinových obsazeností ke snížení počtu qubitů. Pro molekulu předpokládáme, že vzdálenost mezi dvěma atomy vodíku je 0.735 .
(Kvantový) Ansatz: zkušební vlnová funkce (jak sestavit jednoduché kvantové stavy pomocí kvantového obvodu)
Ve VQE se ansatz (množné číslo: ansätze) skládá ze dvou klíčových složek. První je příprava počátečního stavu, která nastaví stav qubitu aplikací kvantových hradel bez variačního parametru. Druhou složkou je parametrizovaný kvantový obvod — speciální kvantový obvod s nastavitelnými parametry, podobnými knoflíkům na rádiu. Tyto parametry budou použity v poslední části — klasickém optimalizátoru — aby nám pomohly dosáhnout nejlepšího možného základního stavu.
V části o variačním principu jsme se dozvěděli, že kvalita zkušebního stavu ovlivňuje kvalitu výsledků variačního algoritmu. To znamená, že volba dobrého ansatzu je ve VQE důležitá. Opět se jedná o bohaté a složité téma. Různé typy ansatzu ani jejich původ zde nebudeme rozebírat. Pokud tě zajímá více o parametrizovaných kvantových obvodech a ansatzu, můžeš prozkoumat lekci Ansatz and variational form z kurzu Variational algorithm design, která poskytuje podrobná vysvětlení a příklady ansätze.
Protože v tomto modulu budeme používat jednoqubitový hamiltonián, potřebujeme jako ansatz jednoqubitový parametrizovaný kvantový obvod. V následující části uvidíme tři typy jednoqubitových ansätze. Porovnáme je a probereme klíčová hlediska při výběru ansatzu.
(Klasický) Optimalizátor: doladění kvantového obvodu
Jakmile kvantový počítač změří energii observably z ansatzu, parametry ansatzu a hodnota energie jsou odeslány klasickému optimalizátoru k ladění. Tento optimalizační proces probíhá na klasickém počítači, přičemž se typicky využívají obecné vědecké balíčky jako SciPy.
Klasický optimalizátor zachází s naměřenou energií jako s nákladovou funkcí. V optimalizačních problémech je nákladová funkce (někdy také nazývaná účelová funkce) matematická funkce, která měří, jak „dobré" je konkrétní řešení. Cílem optimalizátoru je najít sadu parametrů, která tuto nákladovou funkci minimalizuje. V kontextu hledání energie základního stavu molekuly slouží samotná energie jako nákladová funkce — chceme najít parametry pro náš kvantový obvod (naše „řešení"), které dávají nejnižší možnou energii. Klasický optimalizátor použije tuto naměřenou hodnotu energie (náklady) a určí další sadu optimalizovaných parametrů pro kvantový ansatz. Tyto aktualizované parametry jsou pak odeslány zpět do kvantového obvodu a celý proces se opakuje. Při každé iteraci klasický optimalizátor upravuje parametry, aby se pokusil snížit energii (minimalizovat nákladovou funkci), dokud není splněno předem definované konvergenční kritérium — ideálně tak, aby byla nalezena nejnižší možná energie (odpovídající základnímu stavu molekuly pro danou vzdálenost vazby a bázovou sadu).
Vědecké balíčky jako SciPy nabízejí mnoho optimalizačních strategií. Více najdeš v lekci Optimization loops kurzu Variational algorithm design. Zde budeme používat COBYLA (Constrained Optimization BY Linear Approximations), optimalizační algoritmus vhodný pro složité energetické krajiny. COBYLA zejména nepokouší se vypočítat gradient studované funkce — jde o tzv. optimalizátor bez gradientu. Představ si, že se snažíš najít nejvyšší vrchol v horách se zavřenýma očima. Protože celou krajinu nevidíš, děláš malé kroky různými směry a kontroluješ, jestli stoupáš nebo klesáš. COBYLA funguje podobně — pohybuje se v prostoru parametrů, testuje různé hodnoty a postupně zlepšuje výsledek, dokud nenajde ten nejlepší.
Nyní jsi připraven(a) provést výpočet VQE. Zkus níže uvedenou kontrolní otázku, která shrnuje celý proces.
Ověř si porozumění
Doplň prázdná místa správnými pojmy tak, aby byl souhrn procesu VQE úplný, a klikni pro zobrazení správných odpovědí.
VQE je variační kvantový algoritmus, který kombinuje sílu (1) ________ a klasického počítání a slouží k nalezení (2) __________ molekuly. Proces začíná definováním (3) __________, který představuje celkovou energii systému a funguje jako observabla v kvantových měřeních. Dále připravíme (4) __________, kvantový obvod s nastavitelnými parametry, který představuje zkušební vlnovou funkci molekuly. Tyto parametry jsou optimalizovány pomocí (5) __________, klasického algoritmu, který iterativně upravuje parametry tak, aby minimalizoval naměřenou energii. Ve výše uvedené diskusi jsme použili optimalizátor (6) __________, který zpřesňuje parametry ansatzu bez potřeby výpočtu derivací. Proces pokračuje, dokud nedosáhneme (7) __________, což znamená, že jsme nalezli nejnižší možnou energii molekuly.
Banka slov:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
Answer
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
Výpočet energie základního stavu atomu vodíku pomocí VQE
Nyní využijme to, co jsme se naučili, k výpočtu energie základního stavu atomu vodíku. V celém modulu budeme používat framework pro kvantové počítání známý jako „Qiskit patterns", který rozděluje pracovní postupy do následujících kroků:
- Krok 1: Mapování klasických vstupů na kvantový problém
- Krok 2: Optimalizace problému pro kvantové provedení
- Krok 3: Provedení pomocí primitiv Qiskit Runtime
- Krok 4: Postprocesing a klasická analýza

Tyto kroky budeme obecně dodržovat.
Začněme načtením některých nezbytných balíčků, včetně primitiv Qiskit Runtime. Také vybereme nejméně vytížený dostupný kvantový počítač.
Níže je kód pro uložení přihlašovacích údajů při prvním použití. Po uložení do svého prostředí tyto informace z notebooku určitě smaž, aby nebyly tvé přihlašovací údaje omylem sdíleny při sdílení notebooku. Další pokyny najdeš v sekcích Set up your IBM Cloud account a Initialize the service in an untrusted environment.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
Níže uvedená buňka ti umožní přepínat mezi použitím simulátoru nebo reálného hardwaru v průběhu celého notebooku. Doporučujeme ji spustit nyní:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Krok 1: Namapuj problém na kvantové obvody a operátory
Výpočet VQE zahajujeme definicí Hamiltoniánu pro molekulu vodíku () při konkrétní vzdálenosti vazby. Tento Hamiltonián reprezentuje celkovou energii systému v podobě qubitových operátorů a byl získán a namapován z molekulárního systému standardním postupem: 1) použití báze STO-6G (konkrétní sada matematických funkcí sloužící k aproximaci elektronových orbitalů), 2) aplikace Jordan-Wignerova mapování (technika převodu fermionových operátorů popisujících elektrony na qubitové operátory) a 3) redukce qubitů pomocí parity Hamiltoniánu za účelem zjednodušení problému.
Jak jsme již dříve vysvětlili, vypočtené energie základního stavu silně závisí na výběru báze a molekulární geometrii (například vzdálenosti vazby). Pro tuto konkrétní konfiguraci a po těchto transformacích je výsledný qubitový Hamiltonián jednoduchý:
Zde představuje operátor identity a představuje Pauliho operátor Z, působící na jediný qubit. Koeficienty jsou odvozeny z integrálů vypočtených pomocí báze STO-6G při této konkrétní vzdálenosti vazby se správnou transformací.
S takto definovaným Hamiltonánem můžeme nyní použít VQE k výpočtu energie jeho základního stavu. Je užitečné porovnat naši vypočtenou energii základního stavu s očekávanými hodnotami. Pro jediný izolovaný atom vodíku (H) je energie základního stavu přesně -0,5 Hartree (v nepřítomnosti relativistických efektů). Vypočítejme přesnou energii základního stavu našeho konkrétního qubitového Hamiltoniánu, jak je definován výše, a porovnejme ji se známými relevantními hodnotami.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Dále potřebujeme parametrizovaný kvantový obvod – ansatz – pro přípravu zkušební vlnové funkce pro základní stav. Cílem je najít parametry , které minimalizují střední hodnotu energie . Výběr ansatzu je zásadní, protože určuje množinu kvantových stavů, které náš obvod dokáže připravit. „Dobrý" ansatz je dostatečně flexibilní, aby mohl reprezentovat stav velmi blízký skutečnému základnímu stavu zkoumaného Hamiltoniánu, ale zároveň není tak složitý, že by vyžadoval příliš mnoho parametrů nebo příliš hluboký obvod pro současné kvantové počítače.
Zde vyzkoušíme tři různé jednoqubitové ansätze, abychom zjistili, který z nich poskytuje lepší „pokrytí" možných kvantových stavů, v nichž se může jeden qubit nacházet. „Pokrytím" se rozumí rozsah kvantových stavů, které obvod ansatzu dokáže vytvořit změnou svých parametrů.
Použijeme tři ansätze založené na různých kombinacích jednoqubitových rotačních hradel:
- Ansatz s jednou jednoosou rotací: Tento ansatz využívá rotace pouze kolem jediné osy (). Na Blochově sféře to odpovídá pohybu podél jediné kružnice. Jde o nejméně flexibilní variantu, která pokrývá omezený soubor stavů.
- Dva dvouosé rotační ansätze: Tyto ansätze kombinují rotace kolem dvou různých os ( a ). To umožňuje dosáhnout větší části Blochovy sféry ve srovnání s jednoosou rotací.
Porovnáním výsledků VQE získaných s těmito třemi ansätzy uvidíme, jak flexibilita a pokrytí stavového prostoru ansatzu ovlivňují naši schopnost nalézt skutečnou energii základního stavu našeho zjednodušeného Hamiltoniánu. Flexibilnější ansatz má potenciál nalézt lepší aproximaci, ale může být také náročnější pro klasický optimalizátor.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Nyní vygenerujeme 5 000 náhodných čísel pro každý parametr a vykreslíme rozložení náhodných kvantových stavů, které tři ansätze produkují s těmito náhodnými parametry. Tyto parametry si můžeš představit jako rotace kolem různých os na povrchu koule. Abychom viděli rozložení kvantových stavů, použijeme Blochovu sféru – trojrozměrnou kouli zobrazující stav jednoho qubitu. Každý bod na sféře reprezentuje možný stav qubitu, přičemž severní a jižní pól odpovídají klasickým hodnotám „0" a „1", ale qubit může být kdekoliv mezi nimi a projevovat speciální kvantové vlastnosti, jako je superpozice. Nejprve si připravíme potřebné funkce pro vykreslení 3D Blochovy sféry a vygenerujeme 5 000 náhodných parametrů.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Podívejme se, jak funguje náš první ansatz.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
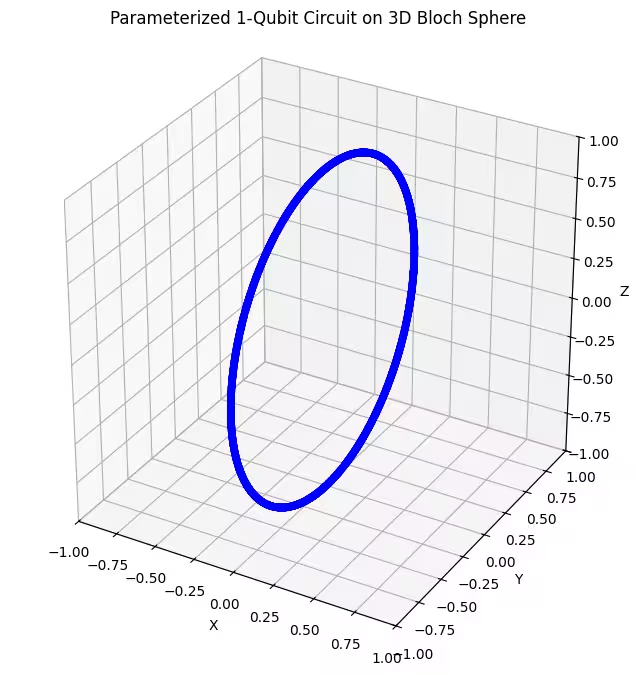
Vidíme, že náš první ansatz vrací prstencově rozložené kvantové stavy na Blochově sféře. To dává smysl, protože jsme ansatzu poskytli pouze jediný rotační parametr. Může tedy produkovat pouze stavy otočené kolem jedné osy. Začínáme-li z bodu a rotujeme kolem jedné osy, vždy dostaneme kružnici. Zkontrolujme nyní náš druhý ansatz, který má dvě ortogonální rotační hradla – Rx a Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
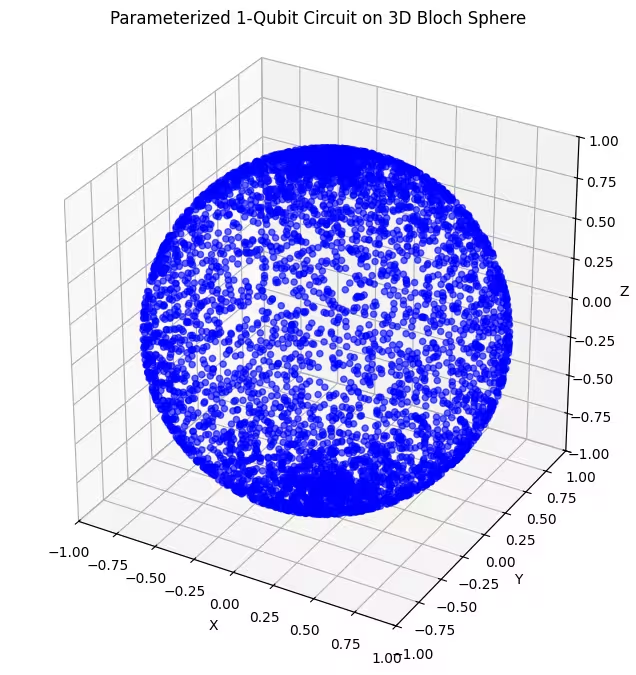
Vidíme, že náš druhý ansatz pokrývá větší část Blochovy sféry – všimni si ale, že body jsou hustěji soustředěny u pólů a řidší u rovníku. Nyní je čas zkontrolovat náš poslední ansatz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
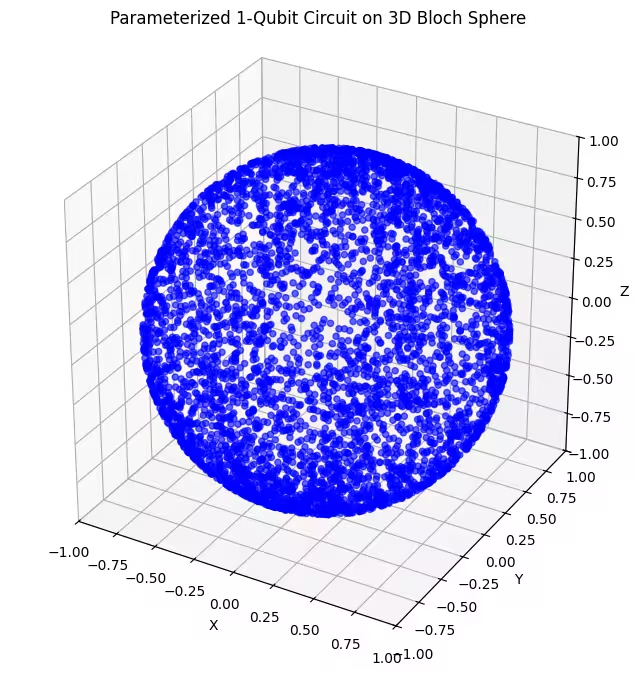
Zde vidíš rovnoměrněji rozložené kvantové stavy generované naším posledním ansatzem.
Jak bylo zmíněno, nejlepší přístup je získat znalosti o hledaném základním stavu a zvolit ansatz, který je vhodný pro zkoumání stavů blízkých tomuto základnímu stavu. Pokud bychom například věděli, že náš základní stav je blízko pólu, mohli bychom zvolit ansatz 2. Pro jednoduchost zůstaneme u ansatzu 3, který rovnoměrně pokrývá celou Blochovu sféru.
Teď, když jsme vybrali náš ansatz, pojďme vykreslit obvod.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Krok 2: Optimalizace pro cílový hardware
Když spouštíme výpočet na skutečném kvantovém počítači, nezáleží nám jen na logice kvantového obvodu. Záleží nám také na tom, jaké operace může daný kvantový počítač vůbec provést a kde na kvantovém počítači se nacházejí qubity, které používáme. Jsou hned vedle sebe? Nebo daleko od sebe? Proto je dalším krokem přepis našeho obvodu pomocí hradel, která jsou pro daný kvantový počítač přirozená, a to s ohledem na rozmístění qubitů. Toho se dosáhne pomocí transpilace – po tomto procesu uvidíš, jak je náš jednoduchý ansatz převeden do jiné sady hradel, a naše abstraktní qubity budou namapovány na fyzické qubity na skutečném kvantovém počítači.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Vidíš, že hradla rx, rz z našeho ansatzu byla převedena na sérii hradel rz, sx, což jsou nativní hradla našeho backendu. Také vidíš, že náš q0 je nyní namapován na pátý fyzický qubit. Musíme také namapovat náš Hamiltonian podle těchto změn, jak ukazuje následující kód:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Krok 3: Spuštění na cílovém hardware
Teď je čas spustit naše VQE na skutečném QPU. K tomu nejprve potřebujeme cenovou funkci pro optimalizační proces, která vyhodnocuje očekávanou hodnotu Hamiltonianu s kvantovým stavem generovaným ansatzem. Nemusíš se bát! Nemusíš psát všechno sám. Připravili jsme pro tebe funkci a vše, co musíš udělat, je spustit níže uvedenou buňku.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Nakonec připravíme počáteční parametry pro náš ansatz a jeho optimalizační proces. Můžeš jednoduše použít samé nuly nebo náhodné hodnoty. Níže jsme vybrali počáteční parametry, ale klidně zakomentuj nebo odkomentuj řádky v buňce, aby se parametry vzorkovaly náhodně, rovnoměrně od 0 do .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Gratulujeme! Právě jsi úspěšně dokončil/a svůj první experiment kvantové chemie. Vidíme rozdíl mezi přesnou energií základního stavu Hamiltonianu a naším výsledkem, ale protože jsme použili výchozí techniku potlačení chyb (která opravuje chyby při čtení), je tento rozdíl malý. To je velmi dobrý začátek!
Poznámka: Lepšího výsledku lze dosáhnout nastavením úrovně potlačení chyb pomocí resilience_level. Výchozí hodnota je 1 a pokud nastavíš vyšší hodnotu, spotřebuje to více času QPU, ale může to vrátit lepší výsledek.
Krok 4: Postprocesing
Nastal čas podívat se na to, jak náš klasický optimalizátor pracoval. Spusť níže uvedenou buňku a sleduj vzor konvergence.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Začali jsme s poměrně dobrou počáteční hodnotou, takže jsme získali dobrý výsledek již za 10 kroků. Vidíš velké i malé vrcholy a to je typická vlastnost optimalizátoru COBYLA – prohledává prostor, jako by neviděl krajinu, a s každým měřením upravuje velikost kroku.
Otestuj si své porozumění
Jaké jsou tvé poznatky? Která část výše uvedeného procesu je otevřena ke zlepšení, aby se dosáhlo výsledků bližších teoretickým hodnotám nebo přesné energii základního stavu Hamiltonianu? Co je třeba vzít v úvahu?
Answer
Prvním aspektem ke zvážení je změna sady bází použitých při výpočtu Hamiltonianu molekul. Jak bylo zmíněno dříve, energie základního stavu atomu H je -0,5 Hartree, což je dobře známý fakt, a báze STO-6G, kterou jsme zvolili, není dostatečná pro přesné odvození této hodnoty.
Volba složitějšího druhu báze zvyšuje počet qubitů použitých Hamiltonianem; proto musíme vybrat složitější a vhodnější ansatz pro chemické problémy.
Dalším aspektem k optimalizaci je správa šumu v QPU. Pokročilejší techniky potlačení chyb přinášejí lepší výsledky, ale jejich použití může trvat déle. Zvažte také, jak shot_number ovlivňuje výsledky.
Nakonec lze lepšího výkonu konvergence dosáhnout také vyzkoušením různých optimalizátorů.
Výpočet energie základního stavu molekuly vodíku pomocí VQE
Nyní, když jsme se podívali na celkový proces VQE na atomech , vypočítáme energii základního stavu molekuly rychleji.
Krok 1: Mapování problému na kvantové obvody a operátory
Zde ti také poskytujeme jednoqubitový Hamiltonian, který používá bázi STO-6G a Jordanovu-Wignerovu transformaci s redukcí qubitů pomocí symetrie Hamiltonianu. Upozorňujeme, že jsme použili atomovou vzdálenost mezi dvěma atomy vodíku 0.735 .
Na rozdíl od výpočtu jediného atomu vodíku (), pro výpočet základního stavu molekuly vodíku () musíme vzít v úvahu také odpudivou sílu působící mezi jádry dvou atomů vodíku, kromě energie spojené s elektronovými orbitaly. V tomto kroku zadáme tuto hodnotu jako konstantu a ve vstupní úloze tuto hodnotu skutečně vypočítáme.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Krok 2: Optimalizace pro cílový hardware
Protože počet qubitů použitých předchozím VQE a Hamiltonianem je stejný jako Backend určený k výpočtu, použijeme stávající ansatz a jeho optimalizovanou podobu.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Krok 3: Spuštění na cílovém hardware
Teď je čas provést výpočty na skutečném QPU. Téměř vše zůstává stejné, ale použijeme vhodný počáteční bod odpovídající Hamiltonianu. Navíc v iterativní části budou některá nastavení Estimatoru — který slouží k výpočtu očekávaných hodnot Hamiltonianu pro ansatz na QPU — nastavena mírně odlišně než v předchozích výpočtech. Tuto změnu si podrobněji rozebereme v kontrolní otázce.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Ačkoli VQE teoreticky poskytuje horní hranici skutečné energie základního stavu, praktické implementace na reálném nebo hlučném simulovaném kvantovém hardware, stejně jako aproximace prováděné při přípravě Hamiltonianu (například volba bázové sady nebo redukce qubitů), mohou vnášet chyby, které někdy vedou k naměřené energii mírně nižší, než je přesná teoretická hodnota nebo konkrétní numerická reference. Přestože se určité chyby vyskytují, výsledky se zdají být uspokojivé — zejména s ohledem na malý počet kroků. Teď dokončeme tento VQE výpočet pohledem na to, jak optimizer pracoval.
Krok 4: Post-processing
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Ověř si znalosti
Vypočítejme energii jaderné repulze molekuly , kterou jsme zahrnuli jako konstantní hodnotu (0,71997 Hartree).

Použij Coulombův zákon a atomové jednotky, abys získal hodnotu v Hartree.
Answer
Protože jsou obě jádra vodíku kladně nabitá, odpuzují se navzájem elektrostatickou silou. Toto odpuzování popisuje Coulombův zákon:
kde je náboj protonu, je permitivita vakua a je vzdálenost mezi oběma jádry, měřená v metrech nebo Bohrových poloměrech v jednotkách joulů (J).
Pro výpočet této energie v Hartree musíme výše uvedenou rovnici převést do systému Atomových jednotek (AU). V AU platí , a Bohrův poloměr () je roven 1 a stává se základní délkovou mírou v AU. S těmito zjednodušeními se Coulombův zákon redukuje na:
kde musí být měřeno v Bohrových poloměrech ().
Pro převod dané vzdálenosti jader v na potřebujeme tento převodní vztah:
takže se stane .
Energie jaderné repulze dané molekuly je tedy
Výpočet reakční energie
Teď využijeme to, co jsme získali! Použil(a) jsi VQE, variační kvantový eigensolver, k výpočtu energie základního stavu atomu a molekuly . Zbývá použít vypočtené hodnoty k získání reakční energie procesu .
Reakční energie je energetická změna, ke které dochází při reakci látek za vzniku nových látek. Představ si, že něco stavíš: někdy je potřeba do toho vložit energii (jako při skládání kostek), a jindy se energie uvolní (jako míč kutálející se z kopce). V chemii reakce buď energii pohlcují (endotermické), nebo uvolňují (exotermické).
Reakční energii procesu lze vypočítat pomocí následujícího vzorce:
Spuštěním buňky níže si to zobrazíme vizuálně. Použijeme přesnou hodnotu energie základního stavu každého Hamiltoniánu a porovnáme reakční energii přesného řešení a výsledků VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Jak je vidět z obrázku, přestože se vyskytují určité chyby, přesná energie základního stavu Hamiltonánů a reakční energie vypočtená pomocí výsledků VQE jsou si podobné a blíží se hodnotě -0,2 Hartree.
Je třeba poznamenat, že reakční energie tohoto procesu má zápornou hodnotu, což znamená, že se při průběhu procesu energie uvolňuje a výsledná molekula má nižší energii než dva jednotlivé atomy.
- Závěr
Shrňme si, co jsme se dosud naučili.
Nejprve jsme se podívali na dvě důležité aproximační techniky potřebné k řešení problémů kvantové chemie: variační princip a volbu bázové sady, které jsou základem VQE. Variační princip jsme prozkoumali ručně výpočtem energie základního stavu jednoduchého harmonického oscilátoru.
Dále jsme prozkoumali VQE, hojně využívaný algoritmus pro výpočet energie základního stavu kvantového systému. Spustili jsme kód pro výpočet energií základního stavu atomárního vodíku () a molekuly vodíku (). Zejména jsme se naučili, že je nutné získat příslušný molekulární Hamiltonián pro daný systém a transformovat ho do podoby spustitelné na kvantovém počítači. Také jsme viděli, že ansatz – parametrizovaný kvantový obvod – je v rámci VQE potřeba k přípravě zkušebních kvantových stavů, a diskutovali jsme o důležitosti výběru vhodné struktury ansatzového obvodu. Dozvěděli jsme se také, že VQE spoléhá na iterativní optimalizační proces využívající klasický počítač, který řídí kvantový obvod k nalezení stavu s nejnižší energií, a viděli jsme, jak tento proces konverguje.
Nakonec jsme použili vypočtené energie základního stavu a získané prostřednictvím VQE k výpočtu reakční energie procesu .
VQE je výkonný kvantový algoritmus pro blízkou budoucnost, je však důležité si uvědomovat jeho omezení. Výkon VQE silně závisí na volbě ansatzu – nalezení efektivně připravitelného ansatzu, který dokáže přesně reprezentovat skutečný základní stav, je pro větší a složitější molekuly stále náročnější. Současný kvantový hardware je navíc náchylný k šumu, což může ovlivnit přesnost výsledků VQE, zejména pro hlubší obvody nebo větší počty qubitů. Navzdory těmto výzvám VQE slouží jako základní algoritmus a probíhající výzkum zkoumá sofistikovanější variační metody a techniky zmírňování chyb s cílem posunout hranice toho, co je možné v kvantové chemii na kvantových počítačích blízké budoucnosti. Například se vyvíjejí algoritmy jako Sample-based Quantum Diagonalization (SQD), které využívají vzorky získané z kvantových obvodů v kombinaci s klasickou diagonalizací v podprostoru ke zlepšení odhadu energie a řešení některých omezení VQE, zejména pokud jde o efektivitu měření a robustnost vůči šumu.
Opakování a otázky
Klíčové pojmy:
- Variační kvantový algoritmus je výpočetní paradigma, ve kterém klasický počítač a kvantový počítač spolupracují na řešení problému.
- V VQE začínáme Hamiltonánem našeho systému a mapujeme ho na qubity pro spuštění na kvantovém počítači. Vybereme parametrizovaný kvantový obvod – ansatz – a provádíme opakovaná měření, přičemž měníme parametry ansatzu, dokud nedosáhneme nejnižší hodnoty energie. Prohledávání prostoru parametrů probíhá pomocí klasického optimalizátoru. K dosažení dobrých výsledků je nutné zvolit správný ansatz a vhodný optimalizátor.
- Reakční energie je celková energetická změna v chemické reakci, určená rozdílem mezi energií reaktantů a produktů.
Pravda/nepravda
- Variační princip říká, že střední hodnota energie pro libovolnou zkušební vlnovou funkci je vždy větší nebo rovna skutečné energii základního stavu.
- Bázová sada je soubor funkcí používaných k aproximaci kvantových vlnových funkcí.
- VQE je kvantový algoritmus používaný k přesnému řešení Schrödingerovy rovnice pro daný Hamiltonán.
- Ve VQE se k přípravě zkušebních vlnových funkcí používá parametrizovaný kvantový obvod (ansatz).
- Volba optimalizátoru ve VQE (například COBYLA, SPSA nebo ADAM) nemá vliv na kvalitu výsledku.
- Qiskitův
Estimatorse používá k přímému výpočtu středních hodnot Hamiltonánů ve VQE.
Otázky s výběrem odpovědi:
- Jaký je účel Hamiltoniánu ve VQE?
- A) Generovat náhodné kvantové stavy
- B) Určovat energii kvantových stavů
- C) Optimalizovat kvantové obvody
- D) Vytvářet provázanost
- Jaký je hlavní cíl algoritmu VQE?
- A) Najít energii základního stavu Hamiltoniánu
- B) Vytvořit provázanost mezi Qubity
- C) Provést Groverovo hledání
- D) Prolomit RSA šifrování
- Kolik kvantových stavů je v tomto notebooku vygenerováno pro porovnání ansatzu?
- A) 100
- B) 1000
- C) 5000
- D) 10 000
- Proč je ve VQE potřeba klasický optimalizátor?
- A) K provádění kvantových měření
- B) Aktualizovat parametry ansatzu za účelem minimalizace energie
- C) K provázání qubitů
- D) K generování kvantové náhodnosti
- Proč je ansatz navržen jako parametrizovaný?
- A) Aby umožnil přípravu kvantových stavů
- B) Aby umožnil prohledávání širokého prostoru kvantových stavů
- C) Aby snížil složitost obvodu
- D) Aby přímo měřil vlastní hodnoty
- Které z následujících tvrzení je nejpřesnější ohledně volby dobrého ansatzu?
- A) Ansatz musí produkovat stavy rovnoměrně rozložené přes Blochovu sféru, jinak selže.
- B) Ansatz by měl být přizpůsoben tvému systému, aby bylo zajištěno, že dokáže generovat stavy blízké základnímu stavu.
- C) Ansatz by měl produkovat náhodné stavy pomocí svých variačních parametrů.
- D) Lepší ansatz má vždy více variačních parametrů.
(Optional) Příloha: Režie optimalizátoru v závislosti na složitosti ansatze
VQE čelí několika dobře známým výzvám[ref 6] a následující jsou spojeny s tím, co jsme se výše naučili.
- Výzvy při výběru ansatze
Výběr správné variační ansatze je inherentně obtížný. Chemicky inspirované ansätze (jako UCCSD) poskytují fyzikální přesnost, ale vyžadují hluboké obvody, zatímco hardwarově efektivní ansätze mají mělčí obvody, ale mohou postrádat fyzikální interpretovatelnost. Mnoho ansätzí navíc zavádí nadměrný počet variačních parametrů, které k zlepšení přesnosti přispívají jen málo, ale výrazně zvyšují obtížnost optimalizace.
- Obtíže s optimalizací
Optimalizační krajina VQE může obsahovat oblasti, kde gradienty exponenciálně mizí (takzvané „barren plateaus" — pusté plošiny), což klasickým optimalizátorům znesnadňuje efektivní aktualizaci variačních parametrů. Za tímto účelem vyzkoušeli výzkumníci různé typy optimalizátorů — gradientní i bezgradientní — přičemž oba přístupy narážejí na problémy. Gradientní optimalizátory trpí „barren plateaus", zatímco bezgradientní metody vyžadují velký počet vyhodnocení funkce.
- Režie optimalizátoru
Další dobře známou výzvou je režie optimalizátoru, která se vztahuje k rozsahu daného problému. Kvantové obvody potřebné pro VQE rostou do hloubky a složitosti s tím, jak se zvětšuje velikost problému; to obvykle také zvyšuje počet parametrů, které je třeba optimalizovat. S rostoucím počtem parametrů se optimalizační proces stává nezvladatelným, což vede k pomalé konvergenci a obtížím při hledání optimálního řešení.
Zde se na tyto výzvy podíváme pomocí VQE pro molekulu se dvěma různými typy ansätzí.
(Poznámka: Toto může vyžadovat více QPU času, takže pokud nemáš dostatek času, klidně použij simulátor.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Nyní spustíme VQE s počátečním bodem složeným ze samých jedniček, s maximálně 20 kroky, a porovnáme konvergenci obou běhů.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

Výše uvedený graf jasně ukazuje, že optimalizační proces ansatze s více proměnnými potřebuje více času, aby dosáhl stabilní konvergence.
Místo spoléhání se na jednoduché jednoqubitové obvody a přímočarou ansatz se složitost optimalizace zvyšuje, jakmile jsou potřeba větší kvantové obvody a strukturovaně složitější ansätze. To poukazuje na dobře známou výzvu VQE: režii optimalizátoru.
Výzkumníci neustále vyvíjejí různé pokročilé metodologie, které dokážou využít kvantové počítače pro chemické problémy. Různorodé vzdělávací materiály najdeš na IBM Quantum Learning.
Reference
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)