Zlepšení odhadu energie chemického Hamiltoniánu pomocí SQD
V tomto tutoriálu implementujeme Qiskit pattern, který ukazuje, jak post-procesovat zašuměné kvantové vzorky a najít aproximaci základního stavu chemického Hamiltoniánu: molekuly v rovnovážné poloze v bázi 6-31G. Budeme sledovat přístup kvantové diagonalizace na základě vzorků (SQD) ke zpracování vzorků odebraných z 36-qubitového kvantového obvodového ansatzu (v tomto případě obvod LUCJ). Aby se zohlednil vliv kvantového šumu, je použita technika obnovy konfigurací.
Pattern lze popsat ve čtyřech krocích:
- Krok 1: Mapování na kvantový problém
- Generování ansatzu pro odhad základního stavu
- Krok 2: Optimalizace problému
- Transpilace ansatzu pro Backend
- Krok 3: Spuštění experimentů
- Odběr vzorků z ansatzu pomocí primitivu
Sampler
- Odběr vzorků z ansatzu pomocí primitivu
- Krok 4: Post-processing výsledků
- Smyčka sebekonsistentní obnovy konfigurací
- Post-procesování celé sady vzorků bitových řetězců s využitím předchozí znalosti počtu částic a průměrného obsazení orbitalů vypočteného v nejnovější iteraci.
- Pravděpodobnostní vytváření dávek podvzorků z obnovených bitových řetězců.
- Projekce a diagonalizace molekulárního Hamiltoniánu přes každý vzorkovaný podprostor.
- Uložení minimální energie základního stavu nalezené přes všechny dávky a aktualizace průměrného obsazení orbitalu.
- Smyčka sebekonsistentní obnovy konfigurací
Pro tento příklad má Hamiltonián interagujících elektronů obecný tvar:
/ jsou fermionské operátory tvorby/anihilace přidružené k -tému prvku bázové sady a spinu . a jsou jedno- a dvou-tělesové elektronové integrály.
Pracovní postup SQD se sebekonsistentní obnovou konfigurací je znázorněn v následujícím diagramu.
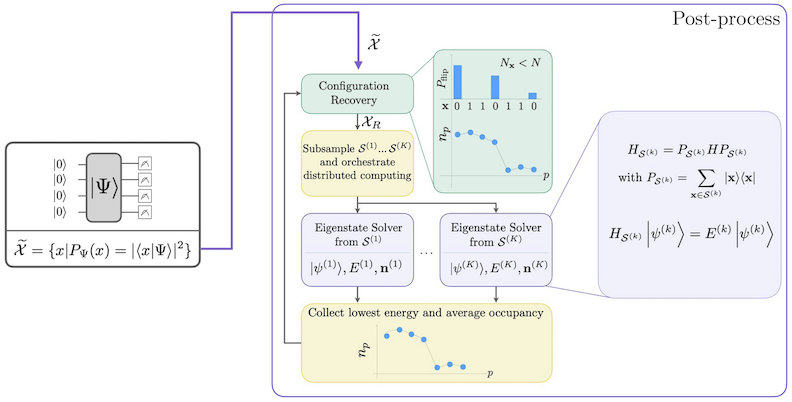
Je známo, že SQD funguje dobře, když je cílový vlastní stav řídký: vlnová funkce je podpořena sadou bázových stavů , jejichž velikost neroste exponenciálně s velikostí problému. V tomto scénáři diagonalizace Hamiltoniánu promítnutého do podprostoru definovaného :
poskytuje dobrou aproximaci cílového vlastního stavu. Úlohou kvantového zařízení je produkovat pouze vzorky členů . Nejprve kvantový Circuit připraví stav v kvantovém zařízení. Je použito Jordanovo-Wignerovo kódování. Členy výpočetní báze tedy představují Fockovy stavy (elektronické konfigurace/determinanty). Circuit je vzorkován ve výpočetní bázi, čímž vznikne sada zašuměných konfigurací . Konfigurace jsou reprezentovány bitovými řetězci. Sada je poté předána do bloku klasického post-processingu, kde je použita technika sebekonsistentní obnovy konfigurací. V rámci SQD je úlohou kvantového zařízení produkovat rozdělení pravděpodobnosti.
Krok 1: Mapování problému na kvantový Circuit
V tomto tutoriálu budeme aproximovat energii základního stavu molekuly . Nejprve specifikujeme molekulu a její vlastnosti. Poté vytvoříme ansatz lokálního unitárního klastrového Jastrowova operátoru (LUCJ) (kvantový Circuit) pro generování vzorků z kvantového počítače k odhadu energie základního stavu.
Nejprve specifikujeme molekulu a její vlastnosti.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Dále vytvoříme ansatz. Ansatz LUCJ je parametrizovaný kvantový Circuit, který inicializujeme amplitudami t2 a t1 získanými z výpočtu CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Použijeme balíček ffsim k vytvoření a inicializaci ansatzu s amplitudami t2 a t1 vypočtenými výše. Protože naše molekula má uzavřenoslupinový stav Hartreeho-Focka, použijeme spin-vyváženou variantu ansatzu UCJ, UCJOpSpinBalanced.
Jelikož náš cílový hardware IBM má topologii těžkého hexagonu, přijmeme vzor zig-zag pro interakce Qubitů. V tomto vzoru jsou orbitaly (reprezentované Qubity) se stejným spinem propojeny lineární topologií (červené a modré kruhy), kde každá linie má tvar zig-zag kvůli konektivitě těžkého hexagonu cílového hardwaru. Opět kvůli topologii těžkého hexagonu mají orbitaly různých spinů propojení každý 4. orbital (0, 4, 8 atd.) (fialové kruhy).
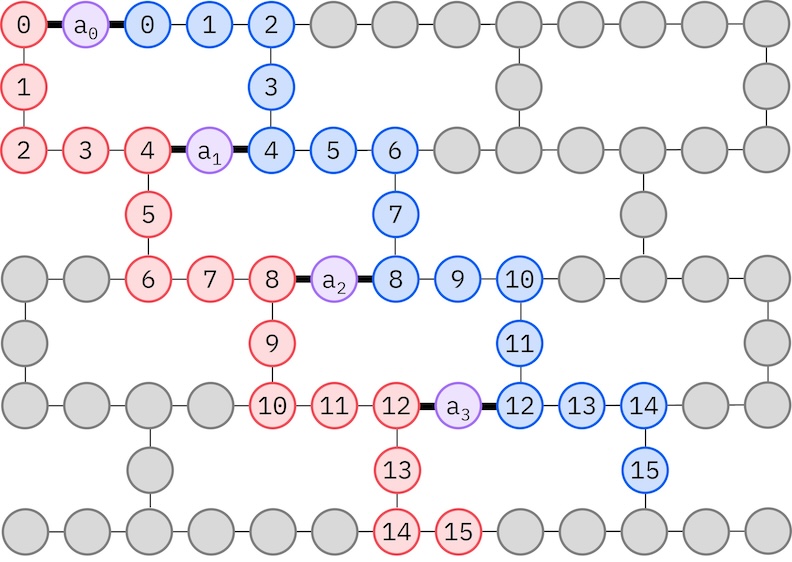
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Krok 2: Optimalizace problému
Dále optimalizujeme náš obvod pro cílový hardware. Před optimalizací Circuit musíme zvolit hardwarové zařízení. Použijeme falešný 127-qubitový Backend z qiskit_ibm_runtime pro emulaci reálného zařízení. Pro spuštění na skutečném QPU jednoduše nahraď falešný Backend skutečným. Více informací najdeš v dokumentaci Qiskit IBM Runtime.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Dále doporučujeme následující kroky pro optimalizaci ansatzu a jeho zpřístupnění pro hardware.
- Vyber fyzické Qubity (
initial_layout) z cílového hardwaru, které odpovídají výše popsanému vzoru zig-zag. Rozložení Qubitů v tomto vzoru vede k efektivnímu obvodu kompatibilnímu s hardwarem s méně hradly. - Vygeneruj staged pass manager pomocí funkce generate_preset_pass_manager z Qiskitu s tvou volbou
backendainitial_layout. - Nastav fázi
pre_inittvého staged pass manageru naffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITobsahuje průchody Transpileru Qiskitu, které rozloží hradla na orbitální rotace a poté tyto orbitální rotace sloučí, což vede k méně hradlům ve výsledném Circuit. - Spusť pass manager na svém Circuit.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Krok 3: Spuštění experimentů
Po optimalizaci Circuit pro spuštění na hardwaru jsme připraveni jej spustit na cílovém hardwaru a sbírat vzorky pro odhad energie základního stavu. Protože máme pouze jeden obvod, použijeme režim spuštění Job Qiskit Runtime a spustíme náš obvod.
Poznámka: Kód pro spuštění Circuit na QPU jsme zakomentovali a ponechali jej jako referenci pro uživatele. Místo spuštění na skutečném hardwaru v tomto průvodci jednoduše vygenerujeme náhodné vzorky z rovnoměrného rozdělení.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Krok 4: Post-processing výsledků
Nyní spustíme algoritmus SQD pomocí funkce diagonalize_fermionic_hamiltonian. Vysvětlení argumentů této funkce najdeš v dokumentaci API.
Solver zahrnutý v addonu SQD používá implementaci selected CI od PySCF, konkrétně pyscf.fci.selected_ci.kernel_fixed_space. Níže uvedený příklad také ukazuje, jak předat klíčové argumenty této funkci prostřednictvím zahrnutého solveru. Zde předáváme argument max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
Nyní zobrazíme výsledky.
První graf ukazuje, že po několika iteracích odhadujeme energii základního stavu s přesností ~16 mH (chemická přesnost je typicky přijímána jako 1 kcal/mol 1.6 mH). Pamatuj, kvantové vzorky v této ukázce byly čistý šum. Signál zde pochází z a priori znalosti elektronické struktury a molekulárního Hamiltoniánu.
Druhý graf ukazuje průměrné obsazení každého prostorového orbitalu po poslední iteraci. Vidíme, že elektrony se spinem nahoru i dolů obsazují s vysokou pravděpodobností prvních pět orbitalů v našich řešeních.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha
