HI-VQE Chemistry – Qiskit Function od Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Viz referenci API
Qiskit Functions jsou experimentální funkce dostupné pouze uživatelům plánů IBM Quantum® Premium, Flex a On-Prem (prostřednictvím IBM Quantum Platform API). Jsou ve stavu preview verze a mohou se měnit.
Package versions
The code on this page was developed using the following requirements. We recommend using these versions or newer.
qiskit-ibm-runtime~=0.45.0
Přehled
V kvantové chemii se problém elektronové struktury zaměřuje na hledání řešení elektronové Schrödingerovy rovnice – kvantových vlnových funkcí popisujících chování elektronů v systému. Tyto vlnové funkce jsou vektory komplexních amplitud, přičemž každá amplituda odpovídá příspěvku jedné možné konfigurace elektronů.
Základní stav je vlnová funkce systému s nejnižší energií a má zvláštní důležitost při studiu molekulárních systémů. Nejpřesnější přístup k výpočtu základního stavu uvažuje všechny možné konfigurace elektronů, ale pro větší systémy se stává nezvládnutelným, protože počet konfigurací roste exponenciálně s velikostí systému.
Handover Iterative Variational Quantum Eigensolver (HI-VQE) je inovativní hybridní kvantově-klasická metoda pro přesný odhad základního stavu molekulárních systémů. Integruje kvantový hardware s klasickými počítači – kvantové procesory efektivně prozkoumávají kandidátní konfigurace elektronů a výsledná vlnová funkce se počítá na klasických počítačích. Díky generování kompaktních, ale chemicky přesných vlnových funkcí HI-VQE podporuje výzkum a objevy v kvantové chemii a vědě o materiálech.

HI-VQE snižuje výpočetní složitost problému elektronové struktury tím, že efektivně odhaduje základní stav s vysokou přesností. Zaměřuje se na pečlivě vybranou podmnožinu nejrelevantnějších konfigurací elektronů, čímž optimalizuje jak přesnost, tak efektivitu.
Kombinací silných stránek klasických i kvantových počítačů HI-VQE iterativně zpřesňuje a vylepšuje aktuální odhad vlnové funkce. Jedinečné techniky konstrukce podprostoru pomáhají zefektivnit výběr konfigurací, takže uživatelé mají větší výpočetní kontrolu a lepší přesnost při simulacích kvantové chemie.
Pokud se chceš dozvědět více o algoritmu do hloubky, můžeš si přečíst příslušný výzkumný článek.
Popis
Počet konfigurací elektronů pro molekulární systém roste exponenciálně s velikostí systému. U určitých elektronových stavů, jako je základní stav, je však běžné, že pouze malá část konfigurací významně přispívá k energii stavu. Metody vybrané konfigurační interakce (SCI) využívají tuto řídkost ke snížení výpočetních nákladů tím, že identifikují a zaměřují se na nejrelevantnější konfigurace. Tato podmnožina konfigurací se označuje jako podprostor.
HI-VQE využívá přirozenou efektivitu kvantových počítačů při reprezentaci molekulárních systémů k usnadnění hledání podprostoru. Integruje klasické a kvantové podrutiny k řešení problému elektronové struktury s vysokou přesností. Na rozdíl od stávajících kvantových SCI metod HI-VQE kombinuje variační trénink, iterativní konstrukci podprostoru a předdiagonalizační filtrování konfigurací, čímž zvyšuje efektivitu snížením počtu kvantových měření, iterací a nákladů na klasickou diagonalizaci. HI-VQE lze proto aplikovat na větší molekulární systémy vyžadující více qubitů a snižuje náklady na řešení problému dané velikosti na stejnou úroveň přesnosti.
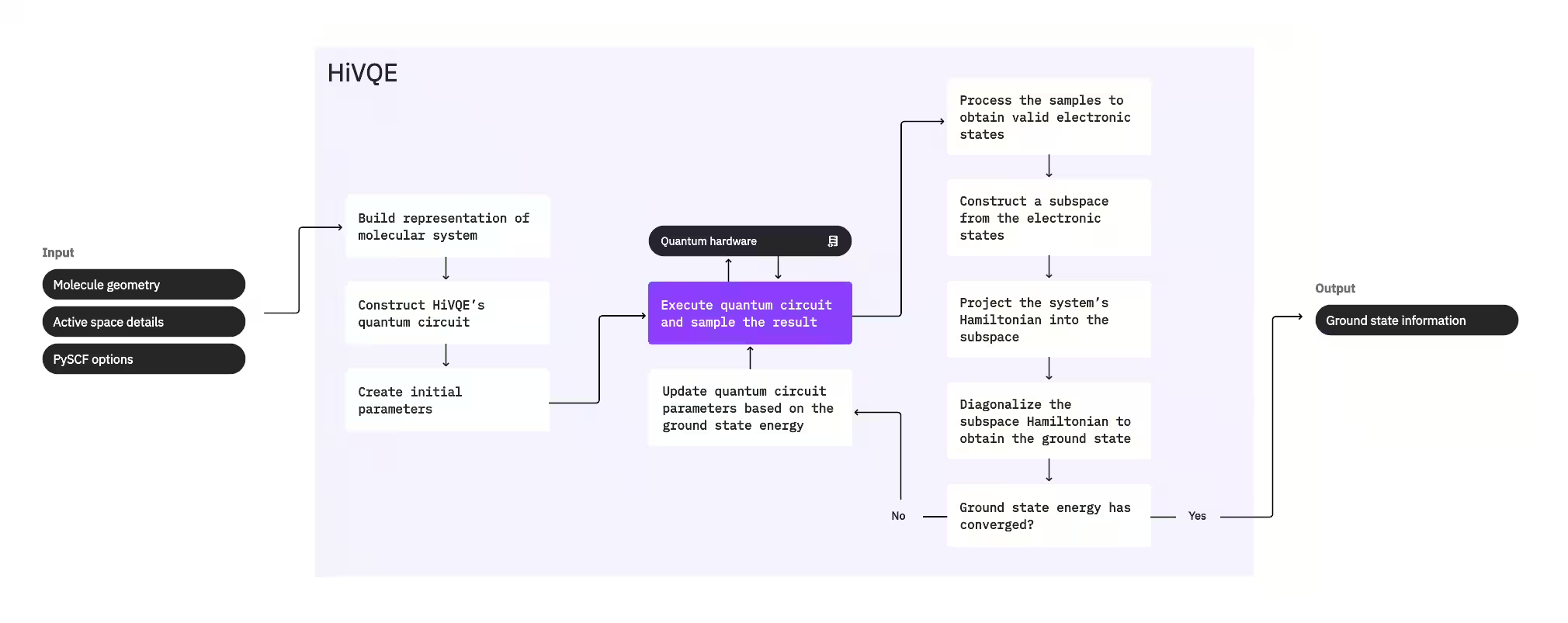
Pro výpočet základního stavu systému HI-VQE nejprve použije klasický chemický balíček PySCF k vygenerování molekulární reprezentace ze vstupů zadaných uživatelem, jako je geometrie molekuly a další molekulární informace. Poté vstoupí do hybridní kvantově-klasické optimalizační smyčky, která iterativně zpřesňuje podprostor tak, aby optimálně reprezentoval základní stav a zároveň minimalizoval počet zahrnutých konfigurací. Smyčka pokračuje, dokud nejsou splněna konvergenční kritéria, jako je velikost podprostoru nebo stabilita energie, načež jsou výstupem vypočítaná vlnová funkce základního stavu a energie. Tyto výsledky lze použít ke konstrukci přesných povrchů potenciální energie a k dalšímu rozboru systému.
Optimalizační smyčka se zaměřuje na úpravu parametrů kvantového Circuit tak, aby generoval vysoce kvalitní podprostor. HI-VQE nabízí tři možnosti kvantového Circuit: excitation_preserving, efficient_su2 a LUCJ. Optimalizace je inicializována blízko referenčního stavu Hartree-Fock pro jeho obecnou vhodnost. Circuit je poté spuštěn na kvantovém zařízení a konfigurace jsou vzorkovány z výsledného kvantového stavu a vráceny jako binární řetězce. Kvůli šumu kvantového zařízení mohou být některé vzorkované konfigurace fyzikálně neplatné, pokud nesplňují zachování počtu elektronů nebo spinu. HI-VQE to řeší pomocí procesu obnovy konfigurací z balíčku qiskit-addon-sqd, takže uživatelé mohou neplatné konfigurace buď opravit, nebo je vyřadit.
Platné konfigurace poté procházejí volitelným filtrovacím krokem, který odstraní ty, u nichž se předpokládá minimální příspěvek. Tím se snižuje dimenze podprostoru, a tedy i náklady na krok diagonalizace. Pokud je filtrování povoleno, vytvoří se předběžný subprostorový Hamiltonián z platných konfigurací a provede se diagonalizace s velmi volnými ukončovacími kritérii. Ačkoli je přesnost výsledných amplitud pro každou konfiguraci nízká, je efektivní pro předpovězení, které konfigurace v dané iteraci z podprostoru vyřadit, a je rychlá na výpočet.
Vybrané konfigurace jsou přidány do podprostoru a Hamiltonián systému je promítnut do tohoto podprostoru. Podprostor se iterativně aktualizuje a zachovává nejrelevantnější konfigurace napříč iteracemi. Tento přístup se liší od alternativních metod tím, že kvantový Circuit nemusí v každém kroku aproximovat celý základní stav.
Následně je subprostorový Hamiltonián klasicky diagonalizován, aby bylo získáno nejnižší vlastní číslo a odpovídající vlastní vektor, reprezentující aproximaci základního stavu a jeho energie. Jak se kvalita podprostoru prostřednictvím iterací zlepšuje, vypočítaný základní stav lépe aproximuje skutečný základní stav. V tomto bodě lze provést dodatečný filtrovací krok k odstranění konfigurací z podprostoru, které významně nepřispívají k vypočítanému základnímu stavu. Tento krok zajišťuje, že podprostor přenesený do další iterace je co nejkompaktnější. Je vyhodnocován na základě amplitud vrácených diagonalizací, protože ty reprezentují příspěvek každé konfigurace k vypočítanému základnímu stavu.
Kontrola konvergence pak určí, zda by další trénink zlepšil výsledky. Pokud ano, provede se volitelný krok klasické expanze, parametry kvantového Circuit se aktualizují pro další minimalizaci vypočítané energie a proces se opakuje. Krok klasické expanze generuje další konfigurace pro podprostor, doplňující konfigurace vzorkované z kvantového zařízení. Nejprve identifikuje konfiguraci s největší amplitudou ve výsledcích diagonalizace a poté generuje nové konfigurace s jednoduchými a dvojnásobnými excitacemi z identifikované konfigurace. Požadovaný počet těchto konfigurací je poté přidán do podprostoru.
Jakmile je určeno, že iterace konvergovaly, HI-VQE vrátí vypočítaný základní stav (ve formě stavů v podprostoru a jejich amplitud ve vlnové funkci základního stavu), jeho energii a míru rozptylu energie, která ukazuje, zda vypočítaný stav tvoří vlastní stav Hamiltoniánu systému.
Uživatelé mohou rozhodovat o tom, který kvantový Circuit se použije, a o počtu shotů pro každý kvantový Circuit, a také řídit velikost podprostoru nebo povolit klasické generování dalších konfigurací na podporu konfigurací generovaných kvantově. Tímto způsobem mohou uživatelé přizpůsobit chování HI-VQE svým požadovaným aplikacím.
Licencování
Vezmi prosím na vědomí, že použití této Qiskit Function je omezeno na problémy vyžadující nejvýše 20 qubitů, pokud není získána licence umožňující vyšší limit.
Pokud máš zájem o získání licence, pošli e-mail na adresu qiskit.support@qunovacomputing.com.
Začínáme
Nejprve si požádej o přístup k funkci. Poté se autentizuj pomocí svého IBM Quantum® API klíče a za předpokladu, že jsi již uložil/a svůj účet do lokálního prostředí, vyber Qiskit Function takto:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Příklad
První příklad ukazuje, jak vypočítat energii základního stavu molekuly NH3 pomocí algoritmu HI-VQE.
Definice molekulární geometrie a možností
Molekulární geometrie NH3 je zadána v kartézských souřadnicích, kde jsou atomy odděleny znakem „;".
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Další možnosti pro molekulární systém lze definovat a zadat v následujícím formátu slovníku.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Spusť funkci se zadanými vstupy geometrie a možností.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Je dobré vytisknout ID úlohy funkce, aby ho bylo možné uvést v žádostech o podporu v případě problémů.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Tento příklad využívá 16 qubitů s 8 orbitaly báze sto3g pro molekulu NH3. Zkontroluj stav svého pracovního zatížení Qiskit Function nebo načti výsledky takto:
print(job.status())
QUEUED
Po dokončení úlohy lze výsledky získat pomocí instance result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Pro přístup k energii základního stavu použij klíč "energy". Klíč "eigenvector" poskytuje CI koeficienty s odpovídající zápisem bitového řetězce konfigurace elektronů uloženého v "states" výsledků.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Výkon
Tato sekce ukazuje provedené referenční výpočty HI-VQE pro případ s 24 qubity pro Li2S, případ s 40 qubity pro molekulu N2 a případ s 44 qubity pro systém FeP-NO.
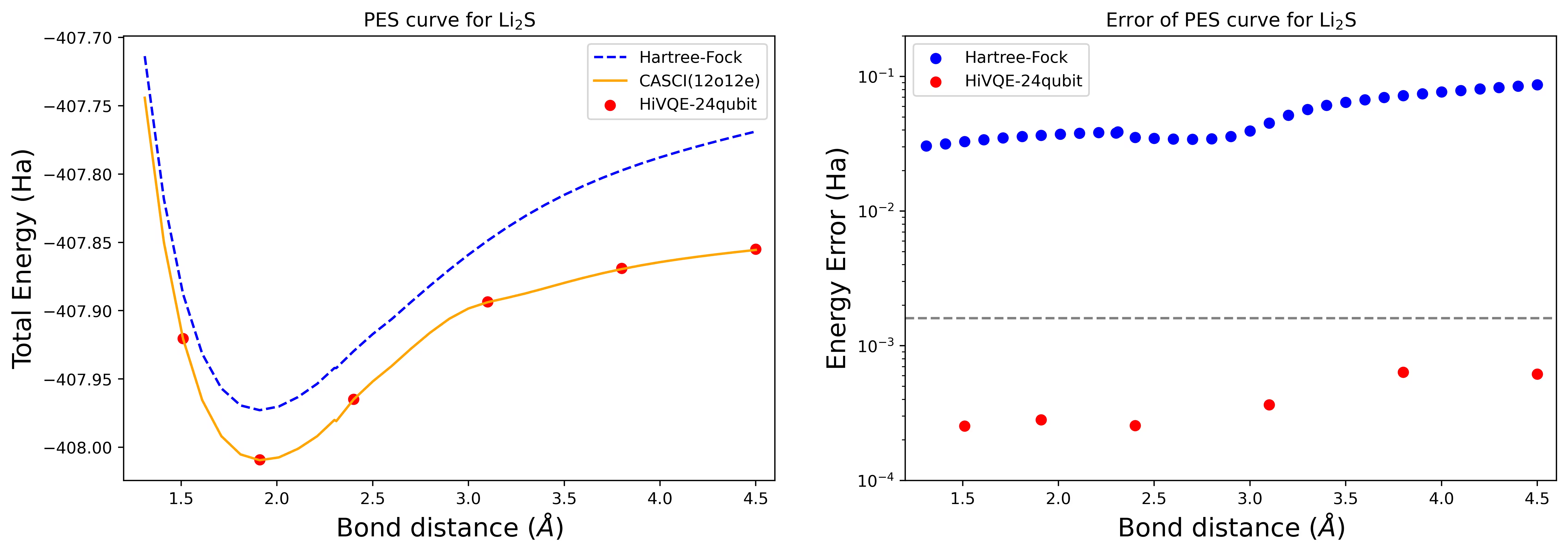
Křivka potenciální energetické plochy disociace molekuly Li2S s 24 qubity
Křivka PES je zobrazena s referenčními hodnotami FCI a počátečním odhadem z RHF spolu s energetickou chybou oproti referenční hodnotě FCI.
 .
.
Výpočty byly provedeny s následujícími geometriemi a možnostmi.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Červené tečky představují výsledky výpočtu HI-VQE pro šest různých geometrií; tři geometrie odpovídající vzdálenostem 1,51, 2,40 a 3,80 Ångströmu jsou zadány jako vstup v buňce výše.
Křivka PES disociace molekuly N2 s 40 qubity
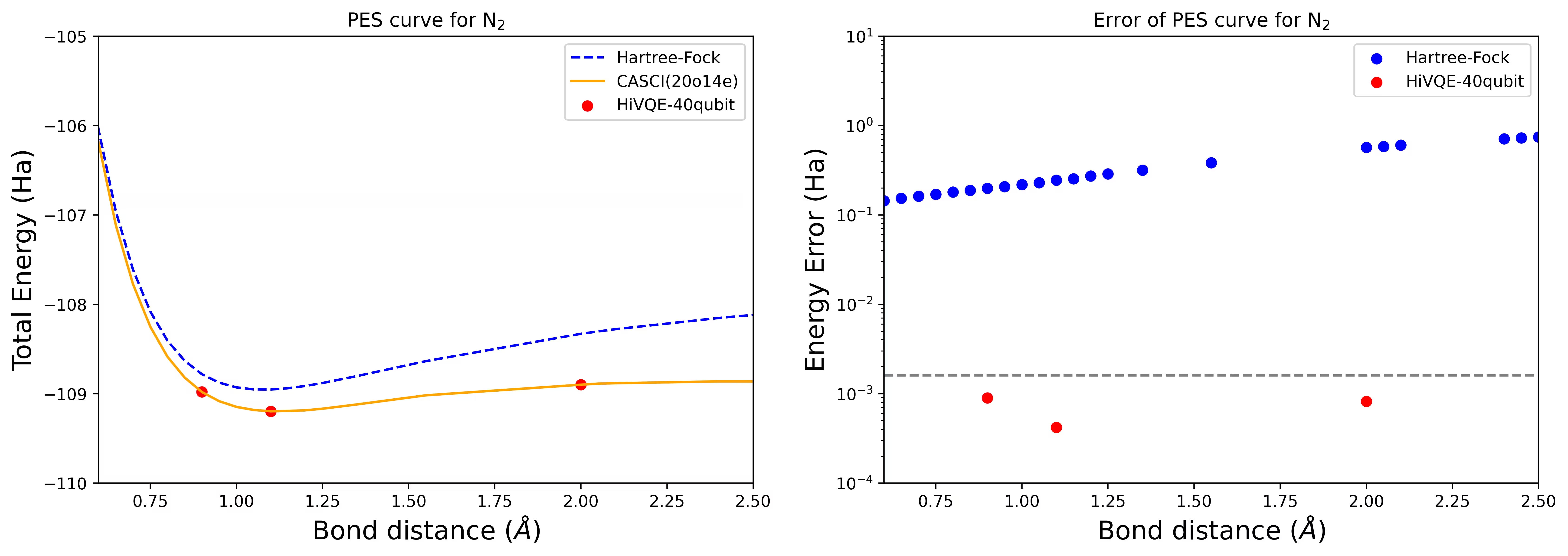
Molekula dusíku byla identifikována jako multireferenční systém s velkými příspěvky korelační energie přesahujícími stav Hartree-Fock. Provedli jsme referenční výpočet molekuly N2 s bází cc-pvdz, (20o,14e) s využitím výběru aktivních orbitalů homo-lumo. Velikost prostoru úplné aktivní vrstvy (CAS) pro popis tohoto problému je 6 009 350 400. Řešení problému vlastních hodnot (pro energii a elektronovou strukturu) s tímto počtem stavů není možné získat na výkonném stolním počítači (16 cpu/64 GB). S HI-VQE mohou uživatelé efektivně prohledávat podprostor stavů CAS a nacházet chemicky přesné výsledky při výrazné úspoře výpočetních zdrojů. Následující grafy ukazují křivku PES výpočtu HI-VQE disociace molekuly N2 s 40 qubity.
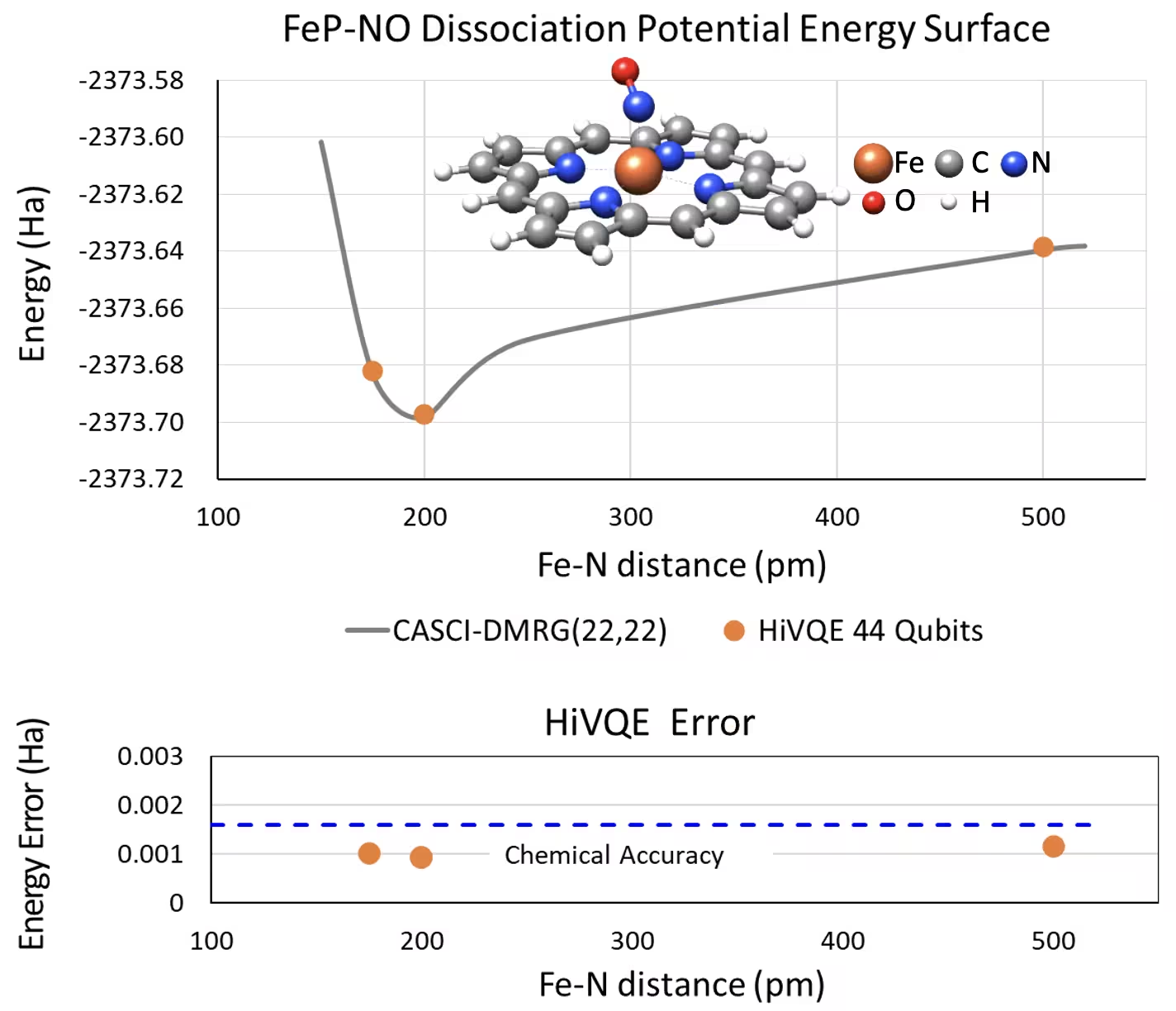
Křivka PES disociace pětisouřadnicového železo(II)-porfyrinu se systémem NO s 44 qubity
Dalším zajímavým chemickým systémem je komplex železo(II)-porfyrin (FeP) s koordinovaným ligandem oxidu dusnatého (NO), který představuje biologicky relevantní metaloporfyrinový systém sehrávající klíčovou roli v různých fyziologických procesech. V tomto příkladu byl HI-VQE využit k odhadu přesné křivky potenciální energetické plochy mezimolekulární interakce mezi FeP a NO (energie základního stavu pro různě vzdálené geometrie). Kombinovaný systém má 450 orbitalů a 202 elektronů (450o,202e) s bází 6-31g(d) celkem. Výběr aktivních orbitalů homo-lumo byl použit k výpočtu zmenšeného případu z reálného případu s (22o,22e). Z následujících referenčních výsledků se nám podařilo dosáhnout chemické přesnosti (> 1,6 mHa) ve srovnání s referenčním výpočtem CASCI(DMRG) (22o,22e) na nejmodernějším klasickém počítači pro chemii.
Benchmarky
- Přesná velikost matice je počet determinantů pro přesné řešení, například FCI a CASCI.
- HI-VQE vzorkuje a počítá jejich podprostor (tj. velikost matice HI-VQE).
- Celkový čas zahrnuje běh na QPU a spuštění Qiskit Function s CPU.
- Přesnost je odhadována z energetického rozdílu oproti přesnému řešení.
| Chemical system | Number of qubits | Exact matrix size | HI-VQE matrix size | E(diff) from exact (mHa) | Number of iteration | Total time | QPU runtime usage |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 s | 258 s |
Načtení chybových zpráv
Pokud tvoje pracovní zatížení selže, stav bude ERROR a volání job.result() vyvolá výjimku:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Získání podpory
Pomoc s touto funkcí můžeš získat zasláním e-mailu na adresu qiskit.support@qunovacomputing.com.
Pokud potřebuješ pomoc s odstraňováním konkrétní chyby, uveď prosím ID úlohy funkce, která chybu způsobila.
Další kroky
- Požádej o přístup k funkci vyplněním tohoto formuláře.
- Navštiv referenci API pro tuto Qiskit Function.
- Vyzkoušej tutoriál Compute dissociation PES curve for FeP-NO with HI-VQE.
- Přečti si Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Vyzkoušej tutoriál Dissociation PES curves with Qunova HiVQE.