Kvantová Krylovova diagonalizace
V této lekci o Krylovově kvantové diagonalizaci (KQD) odpovíme na následující otázky:
- Co je Krylovova metoda obecně?
- Proč Krylovova metoda funguje a za jakých podmínek?
- Jakou roli hraje kvantové počítání?
Kvantová část výpočtů vychází převážně z práce v Ref [1].
Video níže poskytuje přehled Krylovových metod v klasickém počítání, motivuje jejich použití a vysvětluje, jak může kvantové počítání hrát roli v tomto pracovním postupu. Následující text nabízí více podrobností a implementuje Krylovovu metodu jak klasicky, tak na kvantovém počítači.
1. Úvod do Krylovových metod
Krylovova metoda podprostoru může odkazovat na kteroukoli z několika metod postavených kolem toho, čemu se říká Krylovův podprostor. Úplný přehled těchto metod přesahuje rozsah této lekce, ale Ref [2-4] může poskytnout podstatně více pozadí. Zde se zaměříme na to, co Krylovův podprostor je, jak a proč je užitečný při řešení problémů vlastních čísel, a nakonec jak ho lze implementovat na kvantovém počítači. Definice: Je-li dána symetrická, pozitivně semidefinitní matice , pak Krylovův prostor řádu je prostor rozprostřený vektory získanými násobením vyšších mocnin matice , až do , s referenčním vektorem .
Přestože výše uvedené vektory rozprostírají to, co nazýváme Krylovův podprostor, není důvod si myslet, že budou ortogonální. Často se používá iterativní ortonormalizační proces podobný Gram-Schmidtově ortogonalizaci. Zde je tento proces mírně odlišný, protože každý nový vektor je učiněn ortogonálním k ostatním v okamžiku, kdy je generován. V tomto kontextu se tomu říká Arnoldiho iterace. Počínaje počátečním vektorem se generuje další vektor a poté se zajistí, aby byl tento druhý vektor ortogonální k prvnímu odečtením jeho projekce na . To jest
Nyní je snadné vidět, že protože
Totéž provedeme pro další vektor a zajistíme, aby byl ortogonální k oběma předchozím:
Pokud tento proces opakujeme pro všech vektorů, máme úplnou ortonormální bázi pro Krylovův prostor. Všimni si, že ortogonalizační proces zde dá nulový výsledek, jakmile , protože ortogonálních vektorů nutně rozprostírá celý prostor. Tento proces dá také nulový výsledek, pokud je jakýkoli vektor vlastním vektorem , protože všechny následující vektory budou násobky tohoto vektoru.
1.1 Jednoduchý příklad: Krylovova metoda ručně
Pojďme si projít generování Krylovova podprostoru na triviálně malé matici, abychom mohli vidět tento proces. Začneme počáteční maticí , která nás zajímá:
Pro tento malý příklad můžeme určit vlastní vektory a vlastní čísla snadno i ručně. Numerické řešení ukážeme zde.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy sympy
# One might use linalg.eigh here, but later matrices may not be Hermitian. So we use
# linalg.eig in this lesson.
import numpy as np
A = np.array([[4, -1, 0], [-1, 4, -1], [0, -1, 4]])
eigenvalues, eigenvectors = np.linalg.eig(A)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
The eigenvalues are [2.58578644 4. 5.41421356]
The eigenvectors are [[ 5.00000000e-01 -7.07106781e-01 5.00000000e-01]
[ 7.07106781e-01 1.37464400e-16 -7.07106781e-01]
[ 5.00000000e-01 7.07106781e-01 5.00000000e-01]]
Zaznamenáme je zde pro pozdější srovnání:
Rádi bychom prozkoumali, jak tento proces funguje (nebo selhává) s tím, jak zvyšujeme dimenzi našeho Krylovova podprostoru, . Za tímto účelem tento proces aplikujeme:
- Vygenerujeme podprostor celého vektorového prostoru počínaje náhodně zvoleným vektorem (nazveme ho , pokud je již normalizovaný, jak je uvedeno výše).
- Promítneme celou matici na tento podprostor a najdeme vlastní čísla té promítnuté matice .
- Zvětšíme podprostor generováním dalších vektorů, přičemž zajistíme, aby byly ortonormální, pomocí procesu podobného Gram-Schmidtově ortogonalizaci.
- Promítneme na větší podprostor a najdeme vlastní čísla výsledné matice .
- Opakujeme, dokud vlastní čísla nekonvergují (nebo v tomto hračkovém případě dokud nevygenerujeme vektory rozprostírající celý vektorový prostor původní matice ).
Normální implementace Krylovovy metody by nemusela řešit problém vlastních čísel pro matici promítnutou na každý Krylovův podprostor při jeho konstrukci. Mohl bys zkonstruovat podprostor požadované dimenze, promítnout matici na tento podprostor a diagonalizovat promítnutou matici. Promítání a diagonalizace při každé dimenzi podprostoru se provádí pouze za účelem kontroly konvergence.
Dimenze :
Zvolíme náhodný vektor, řekněme
Pokud není již normalizovaný, normalizujeme ho.
Nyní promítneme naši matici na podprostor tohoto jednoho vektoru:
Toto je naše projekce matice na náš Krylovův podprostor, když obsahuje pouze jeden vektor, . Vlastní číslo této matice je triviálně 4. Můžeme to považovat za náš odhad nultého řádu vlastních čísel (v tomto případě pouze jednoho) matice . Přestože je to špatný odhad, je ve správném řádu velikosti.
Dimenze :
Nyní vygenerujeme další vektor v našem podprostoru operací s na předchozím vektoru:
Nyní odečteme projekci tohoto vektoru na náš předchozí vektor, abychom zajistili ortogonalitu.
Pokud není již normalizovaný, normalizujeme ho. V tomto případě byl vektor již normalizovaný, takže
Nyní promítneme naši matici A na podprostor těchto dvou vektorů:
Stále nám zbývá problém určení vlastních čísel této matice. Ale tato matice je o něco menší než plná matice. V problémech zahrnujících velmi velké matice může být práce s tímto menším podprostorem velmi výhodná.
Přestože stále nejde o dobrý odhad, je lepší než odhad nultého řádu. Provedeme tuto iteraci ještě jednou, abychom se ujistili, že je proces jasný. To však podkopává podstatu metody, protože v příští iteraci skončíme diagonalizací matice 3x3, což znamená, že jsme neušetřili čas ani výpočetní výkon.
Dimenze :
Nyní vygenerujeme další vektor našeho podprostoru pomocí operace A na předchozím vektoru:
Nyní odečteme projekci tohoto vektoru na oba předchozí vektory, abychom zajistili ortogonalitu.
Pokud vektor ještě není normalizovaný, normalizuj ho. V tomto případě byl vektor již normalizovaný, takže
Nyní promítneme matici na podprostor těchto vektorů:
Nyní určíme vlastní čísla:
Tato vlastní čísla jsou přesně vlastními čísly původní matice . Tak to musí být, protože jsme rozšířili Krylov podprostor tak, aby pokrýval celý vektorový prostor původní matice .
V tomto příkladu nemusí Krylov metoda vypadat výrazně jednodušeji než přímá diagonalizace. Jak uvidíme v dalších sekcích, Krylov metoda je výhodná teprve od určité dimenze matice; cílem je pomoci nám řešit problémy vlastních čísel a vlastních vektorů pro velmi velké matice.
Toto je jediný příklad, který si ukážeme „ručně", ale sekce 2 níže obsahuje výpočetní příklady.
Vyjasnění pojmů
Běžnou mylnou představou je, že pro daný problém existuje jediný Krylov podprostor. Ale samozřejmě, protože existuje mnoho počátečních vektorů, na které lze naši matici aplikovat, existuje i mnoho možných Krylov podprostorů. Výraz „ten Krylov podprostor" budeme používat pouze tehdy, když odkazujeme na konkrétní Krylov podprostor již definovaný pro konkrétní příklad. Pro obecné přístupy k řešení problémů budeme hovořit o „nějakém Krylov podprostoru". Poslední upřesnění: je platné mluvit o „Krylov prostoru". Často se mu říká „Krylov podprostor" kvůli jeho použití v kontextu promítání matic z počátečního prostoru do podprostoru. V souladu s tímto kontextem budeme o něm zde většinou mluvit jako o podprostoru.
Ověř si své porozumění
Přečti si otázky níže, zamysli se nad svou odpovědí, pak klikni na trojúhelník, aby se zobrazilo řešení.
Vysvětli, proč není (a) užitečné a (b) možné rozšiřovat dimenzi Krylov podprostoru za dimenzi matice, která nás zajímá.
Odpověď:
(a) Protože vektory při jejich generování ortonormalizujeme, vytvoří sada takových vektorů úplnou bázi, což znamená, že jejich lineární kombinací lze vytvořit libovolný vektor v daném prostoru. (b) Ortogonalizační proces spočívá v odečtení projekce nového vektoru na všechny předchozí vektory. Pokud všechny předchozí vektory pokrývají celý vektorový prostor, pak odečtení projekcí na celý podprostor vždy zanechá nulový vektor.
Předpokládejme, že kolega výzkumník předvádí Krylov metodu aplikovanou na malou ukázkovou matici svým kolegům. Tento kolega plánuje použít matici a počáteční vektor:
a
Je na tom něco špatně? Jak bys svému kolegovi poradil/a?
Odpověď:
Tvůj kolega omylem zvolil jako počáteční vektor vlastní vektor. Operace matice na počátečním vektoru vrátí jednoduše stejný vektor zpět, vynásobený vlastním číslem. To nevygeneruje podprostor rostoucí dimenze. Poraď svému kolegovi, aby zvolil jiný počáteční vektor a přesvědčil se, že nejde o vlastní vektor.
Aplikuj Krylov metodu na níže uvedenou matici a zvol vhodný nový počáteční vektor. Zapiš odhady minimálního vlastního čísla na 0. a 1. řádu tvého Krylov podprostoru.
Odpověď:
Existuje mnoho možných odpovědí v závislosti na volbě počátečního vektoru. Zvolíme:
Abychom získali , aplikujeme jednou na a poté uděláme ortogonálním k
Na 0. řádu je projekce na náš Krylov podprostor
Na 1. řádu je projekce na tento Krylov podprostor
To lze provést ručně, ale nejsnáze pomocí numpy:
import numpy as np
vstar = np.array([[1/np.sqrt(3),1/np.sqrt(3),1/np.sqrt(3)],[-1/np.sqrt(6),np.sqrt(2/3),-1/np.sqrt(6)]]
)
A = np.array([[1, 1, 0],
[1, 1, 1],
[0, 1, 1]])
v = np.array([[1/np.sqrt(3),-1/np.sqrt(6)],[1/np.sqrt(3),np.sqrt(2/3)],[1/np.sqrt(3),-1/np.sqrt(6)]])
proj = vstar@A@v
print(proj)
eigenvalues, eigenvectors = np.linalg.eig(proj)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
výstup:
[[ 2.33333333 0.47140452]
[ 0.47140452 -0.33333333]]
The eigenvalues are [ 2.41421356 -0.41421356]
The eigenvectors are [[ 0.98559856 -0.16910198]
[ 0.16910198 0.98559856]]
Odhadované minimální vlastní číslo je -0,414.
1.2 Typy Krylov metod
„Krylov metody podprostorů" mohou odkazovat na kteroukoli z několika iterativních technik používaných k řešení velkých lineárních soustav a problémů vlastních čísel. Co mají všechny společného, je to, že konstruují přibližné řešení z Krylov podprostoru
kde je počáteční odhad (viz ref. [5]). Liší se v tom, jak volí nejlepší aproximaci z tohoto podprostoru, přičemž vyvažují faktory jako rychlost konvergence, využití paměti a celkové výpočetní náklady. Tato lekce se zaměřuje na využití kvantových počítačů v kontextu Krylov metod podprostorů; vyčerpávající diskuze o těchto metodách přesahuje její rozsah. Níže uvedené stručné definice jsou pouze pro kontext a obsahují několik odkazů pro další studium těchto metod.
Metoda sdružených gradientů (CG): Tato metoda se používá k řešení symetrických, pozitivně definitních lineárních soustav[6]. Minimalizuje A-normu chyby v každé iteraci, což ji činí zvláště efektivní pro soustavy pocházející z diskretizovaných eliptických PDR[7]. Tento přístup použijeme v další sekci, abychom motivovali, proč by Krylov podprostor byl efektivním podprostorem pro hledání lepších řešení lineárních soustav.
Metoda zobecněného minimálního rezidua (GMRES): Tato metoda je navržena pro řešení obecných nesymetrických lineárních soustav. Minimalizuje normu rezidua nad Krylov prostorem v každé iteraci, což ji činí robustní, ale potenciálně paměťově náročnou pro velké soustavy[7].
Metoda minimálního rezidua (MINRES): Tato metoda se používá pro řešení symetrických indefinitních lineárních soustav. Je podobná metodě GMRES, ale využívá symetrii matice ke snížení výpočetních nákladů[8].
Mezi další pozoruhodné přístupy patří metoda úplné ortogonalizace (FOM), která úzce souvisí s Arnoldiho metodou pro problémy vlastních čísel, metoda bikonjugovaných gradientů (BiCG) a metoda indukované redukce dimenze (IDR).
1.3 Proč Krylovova metoda podprostoru funguje
Zde si ukážeme, proč by Krylovova metoda podprostoru měla být efektivním způsobem aproximace vlastních hodnot matice prostřednictvím iterativního zpřesňování aproximací vlastních vektorů, a to pohledem přes metodu nejstrmějšího sestupu. Budeme argumentovat tím, že při počátečním odhadu základního stavu tvoří prostor postupných korekcí tohoto počátečního odhadu vedoucích k nejrychlejší konvergenci právě Krylovův podprostor. Rigorizní důkaz konvergenčního chování zde záměrně vynecháváme.
Předpokládejme, že naše matice je symetrická a pozitivně definitní. To činí náš argument nejvíce relevantním pro výše zmíněnou metodu CG. Nepředpokládáme zde nic o řídkosti; ani netvrdíme, že musí být hermitovská (což by musela být, pokud by šlo o Hamiltonian).
Obvykle chceme řešit problém tvaru
Dalo by se uvažovat, že , kde je nějaká konstanta, jako u problému vlastních hodnot. Naše formulace problému ale zatím zůstává obecnější.
Začneme vektorem , který je přibližným řešením. Ačkoli existují paralely mezi tímto odhadem a z oddílu 1.1, zde jich nevyužíváme. Náš odhad má chybu, kterou nazýváme
Dále definujeme reziduum
Zde používáme velké , abychom odlišili reziduum od dimenze našeho Krylovova podprostoru .

Nyní chceme provést korekční krok tvaru
který doufáme zlepší naši aproximaci. Zde je nějaký vektor, který ještě musíme určit. Označme chybu po provedení korekce. Pak

Zajímá nás, jak se chyba chová při transformaci naší maticí. Vypočítejme tedy -normu chyby:
kde jsme využili symetrii a také to, že Zde je nějaká konstanta nezávislá na . Jak bylo zmíněno v oddílu 1.2, -norma chyby není jedinou veličinou, kterou bychom mohli minimalizovat, ale je to dobrá volba. Chceme sledovat, jak se tato veličina mění s naší volbou korekčních vektorů Definujeme proto funkci :
je jen chyba jako funkce korekce měřená v -normě. Chceme tedy zvolit tak, aby bylo co nejmenší. Za tím účelem vypočítáme gradient . S využitím symetrie dostaneme
Gradient ukazuje ve směru nejstrmějšího výstupu, což znamená, že jeho opačný směr nám dává směr, ve kterém funkce klesá nejrychleji: směr nejstrmějšího sestupu. V počátečním odhadu , kde , platí Funkce tedy klesá nejrychleji ve směru rezidua Náš počáteční odhad by tedy nejvíce profitoval z přičtení vektoru pro nějaký skalár .
V dalším kroku opět zvolíme vektor a přičteme jeho hodnotu k aktuální aproximaci. Pomocí stejného argumentu jako dříve volíme pro nějaký skalár . Takto pokračujeme, takže -tá iterace našeho vektoru je
Ekvivalentně chceme budovat prostor, z něhož vybíráme vylepšené odhady, přidáváním , a tak dále v pořadí. -tý odhadovaný vektor leží v
Nyní, s využitím vztahu
vidíme, že
Jinými slovy, prostor, který budujeme a který nejefektivněji aproximuje správné řešení , je přesně prostor vzniklý postupným působením matice na Krylovův podprostor je prostor rozpnutý vektory postupných směrů nejstrmějšího sestupu.
Na závěr zdůrazňujeme, že jsme neučinili žádná numerická tvrzení o škálování tohoto přístupu, ani jsme nediskutovali komparativní výhodu pro řídké matice. Cílem bylo pouze motivovat použití Krylovových metod podprostoru a poskytnout pro ně intuitivní pochopení. Nyní prozkoumáme chování těchto metod numericky.
Ověř své porozumění
Přečti si níže uvedenou otázku, zamysli se nad odpovědí a poté klikni na trojúhelník pro zobrazení řešení.
Ve výše popsaném postupu jsme navrhli minimalizovat -normu chyby. Jaké další veličiny by bylo možné uvažovat při hledání základního stavu a jeho vlastní hodnoty?
Odpověď:
Dalo by se uvažovat o použití vektoru rezidua místo -normy chyby. Mohou existovat případy, kdy je užitečné uvažovat přímo samotný vektor chyby.
2. Krylovovy metody v klasickém výpočtu
V této části implementujeme Arnoldiho iterace výpočetně, abychom mohli využít Krylovův podprostor při řešení problémů vlastních hodnot. Nejprve to aplikujeme na malý příklad, pak prozkoumáme, jak výpočetní čas škáluje s rostoucí velikostí matice. Klíčovou myšlenkou bude, že generování vektorů rozpínajících Krylovův prostor bude významným přispěvatelem k celkové potřebné době výpočtu. Nároky na paměť se liší mezi konkrétními Krylovskými metodami. Ale paměťová omezení mohou limitovat použití tradičních Krylovových metod.
2.1 Jednoduchý příklad v malém měřítku
Při vytváření Krylovova podprostoru budeme muset ortogonalizovat vektory v našem podprostoru. Definujme funkci, která vezme zavedený vektor z našeho podprostoru vknown (bez předpokladu normalizace) a kandidátní vektor vnext, který chceme do podprostoru přidat, a učiní vnext kolmým na vknown a normalizovaným. Dále definujme funkci, která tento postup provede pro všechny zavedené vektory v našem Krylovově podprostoru, čímž zajistíme plně ortonormální sadu.
# vknown is some established vector in our subspace. vnext is one we wish to add,
# which must be orthogonal to vknown.
def orthog_pair(vknown, vnext):
vknown = vknown / np.sqrt(vknown.T @ vknown)
diffvec = vknown.T @ vnext * vknown
vnext = vnext - diffvec
return vnext
# v is the candidate vector to be added to our subspace. s is the existing subspace.
def orthoset(v, s):
v = v / np.sqrt(v.T @ v)
temp = v
for i in range(len(s)):
temp = orthog_pair(s[i], temp)
v = temp / np.sqrt(temp.T @ temp)
return v
Nyní definujme funkci, která iterativně buduje stále větší Krylovův podprostor, dokud prostor Krylovových vektorů nepokryje celý prostor původní matice. To nám umožní sledovat, jak dobře odpovídají vlastní čísla získaná naší metodou Krylovova podprostoru přesným hodnotám, jako funkci dimenze Krylovova podprostoru. Funkce krylov_full_build vrací Krylovovy vektory, promítnuté Hamiltoniany, vlastní čísla a potřebný čas.
# Necessary imports and definitions to track time in microseconds
import time
def time_mus():
return int(time.time() * 1000000)
# This function constructs a Krylov subspace that spans the whole space of the original matrix.
# Input:
# v0 : initial vector
# matrix : original matrix to be diagonalized
# Output:
# ks : Krylov vectors
# Hs : projected Hamiltonians
# eigs : eigenvalues
# k_tot_times : time required for the operation
def krylov_full_build(v0, matrix):
t0 = time_mus()
b = v0 / np.sqrt(v0 @ v0.T)
A = matrix
ks = []
ks.append(b)
Hs = []
eigs = []
Hs.append(b.T @ A @ b)
eigs.append(np.array([b.T @ A @ b]))
k_tot_times = []
for j in range(len(A) - 1):
vec = A @ ks[j].T
ortho = orthoset(vec, ks)
ks.append(ortho)
ksarray = np.array(ks)
Hs.append(ksarray @ A @ ksarray.T)
eigs.append(np.linalg.eig(Hs[j + 1]).eigenvalues)
k_tot_times.append(time_mus() - t0)
# Return the Krylov vectors, the projected Hamiltonians, the eigenvalues,
# and the total time required.
return (ks, Hs, eigs, k_tot_times)
Otestujeme to na matici, která je stále poměrně malá, ale větší, než bychom chtěli zpracovávat ručně.
# Define our small test matrix
test_matrix = np.array(
[
[4, -1, 0, 1, 0],
[-1, 4, -1, 2, 1],
[0, -1, 4, 3, 3],
[1, 2, 3, 4, 0],
[0, 1, 3, 0, 4],
]
)
# Give the test matrix and an initial guess as arguments in the function defined above.
# Calculate outputs.
test_ks, test_Hs, test_eigs, text_k_tot_times = krylov_full_build(
np.array([0.5, 0.5, 0, 0.5, 0.5]), test_matrix
)
Naše funkce můžeme ověřit tak, že v posledním kroku (kdy Krylovův prostor odpovídá celému vektorovému prostoru původní matice) se vlastní čísla z Krylovovy metody přesně shodují s výsledky přesné numerické diagonalizace:
print(np.linalg.eig(test_matrix).eigenvalues)
print(test_eigs[len(test_matrix) - 1])
[-1.36956923 8.43756009 2.9040308 5.34436028 4.68361806]
[-1.36956923 8.43756009 2.9040308 4.68361806 5.34436028]
To se povedlo. Samozřejmě, co skutečně záleží, je kvalita naší aproximace jako funkce dimenze Krylovova podprostoru. Protože nás často zajímá hledání základních stavů a dalších minimálních vlastních čísel (a z dalších algebraických důvodů vysvětlených níže), podívejme se na náš odhad nejmenšího vlastního čísla jako funkci dimenze Krylovova podprostoru. Tedy:
def errors(matrix, krylov_eigs):
targ_min = min(np.linalg.eig(matrix).eigenvalues)
err = []
for i in range(len(matrix)):
err.append(min(krylov_eigs[i]) - targ_min)
return err
import matplotlib.pyplot as plt
krylov_error = errors(test_matrix, test_eigs)
plt.plot(krylov_error)
plt.axhline(y=0, color="red", linestyle="--") # Add dashed red line at y=0
plt.xlabel("Order of Krylov subspace") # Add x-axis label
plt.ylabel("Error in minimum eigenvalue") # Add y-axis label
plt.show()
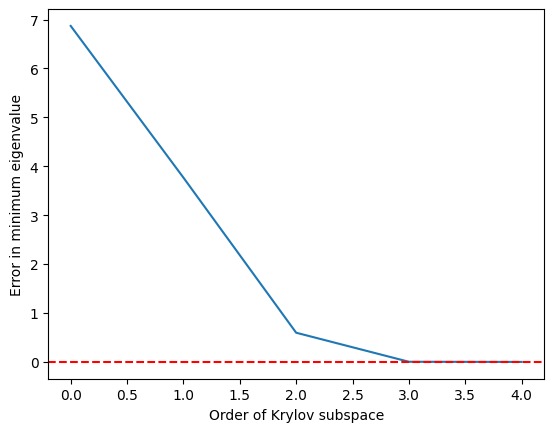
Vidíme, že minimální vlastní číslo je dosaženo poměrně přesně, jakmile Krylovův podprostor vyroste na a je perfektní při
2.2 Časová náročnost v závislosti na dimenzi matice
Přesvědčme se, že Krylovova metoda může být výhodnější než přesné numerické vlastní řešiče, a to takto:
- Sestrojíme náhodné matice (nejsou řídké – to není ideální případ použití pro KQD)
- Určíme vlastní hodnoty dvěma metodami: přímo pomocí NumPy a pomocí Krylovova podprostoru.
- Zvolíme hranici přesnosti, které musí naše vlastní hodnoty dosáhnout, než přijmeme Krylovovy odhady.
- Porovnáme čas potřebný k řešení oběma způsoby.
Výhrady: Jak podrobně probereme níže, Krylovova kvantová diagonalizace se nejlépe hodí pro operátory, jejichž maticové reprezentace jsou řídké nebo je lze zapsat pomocí malého počtu skupin komutujících Pauliho operátorů. Náhodné matice, které zde používáme, tuto podmínku nesplňují. Jsou užitečné pouze pro zkoumání škály, při níž mohou být klasické Krylovovy metody výhodné. Za druhé, při použití Krylovovy metody budeme počítat vlastní hodnoty pro mnoho Krylovových podprostorů různých velikostí. Zaznamenáme čas potřebný pro Krylovův podprostor minimální dimenze, který dosáhne požadované přesnosti pro vlastní hodnotu základního stavu. I to se trochu liší od řešení problému, který je pro přesné vlastní řešiče nezvládnutelný, protože k posouzení potřebné dimenze používáme přesné řešení.
Začneme vygenerováním sady náhodných matic.
import numpy as np
# Set the random seed
np.random.seed(42)
# how many random matrices will we make
num_matrix = 200
matrices = []
for m in range(1, num_matrix):
matrices.append(np.random.rand(m, m))
Každou matici nyní diagonalizujeme přímo pomocí numpy. Pro pozdější porovnání zaznamenáme čas potřebný k diagonalizaci.
matrix_numpy_times = []
matrix_numpy_eigs = []
for mm in range(num_matrix - 1):
t0 = time_mus()
matrix_numpy_eigs.append(min(np.linalg.eig(matrices[mm]).eigenvalues))
matrix_numpy_times.append(time_mus() - t0)
plt.plot(matrix_numpy_times)
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()
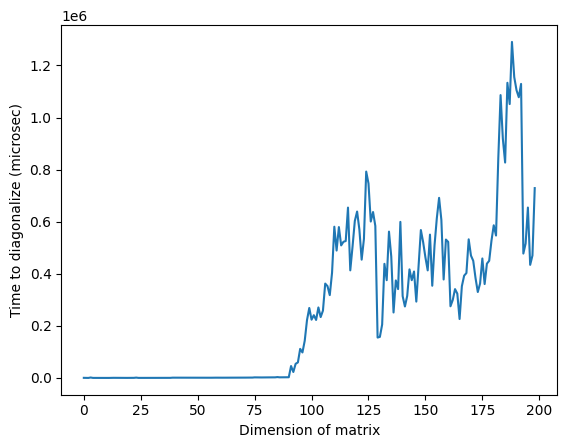
Všimni si, že na obrázku výše může být neobvykle vysoký čas kolem dimenze 125 způsoben náhodnou povahou matic nebo implementací na použitém klasickém procesoru, ale není reprodukovatelný. Opětovné spuštění kódu poskytne jiný profil s různými anomálními špičkami.
Nyní pro každou matici postupně sestavíme Krylovův podprostor a budeme počítat vlastní hodnoty po krocích. V každém kroku zkontrolujeme, zda byla nejnižší vlastní hodnota získána s požadovanou absolutní chybou. Podprostor, který jako první poskytne vlastní hodnoty v rámci zadané chyby, je ten, pro který zaznamenáme časy výpočtu. Provedení této buňky může trvat několik minut v závislosti na rychlosti procesoru. Klidně přeskoč vyhodnocení nebo zmenši maximální dimenzi diagonalizovaných matic. Prohlédnout si předem vypočtené výsledky je dostačující.
# Choose the absolute error you can tolerate, and make a list for tracking the Krylov subspace size
# at which that error is achieved.
abserr = 0.05
accept_subspace_size = []
# Lists to store total time spent on the Krylov method, and the subset of that time spent on
# diagonalizing the projected matrix.
matrix_krylov_tot_times = []
matrix_krylov_dim = []
# Step through all our random matrices
for mm in range(0, num_matrix - 1):
test_ks, test_Hs, test_eigs, test_k_tot_times = krylov_full_build(
np.ones(len(matrices[mm])), matrices[mm]
)
# We have not yet found a Krylov subspace that produces our minimum eigenvalue to
# within the required error.
found = 0
for j in range(0, len(matrices[mm]) - 1):
# If we still haven't found the desired subspace...
if found == 0:
# ...but if this one satisfies the requirement, then record everything
if (
abs((min(test_eigs[j]) - matrix_numpy_eigs[mm]) / matrix_numpy_eigs[mm])
< abserr
):
accept_subspace_size.append(j)
matrix_krylov_tot_times.append(test_k_tot_times[j])
matrix_krylov_dim.append(mm)
found = 1
Vynesme časy, které jsme pro tyto dvě metody získali, pro porovnání:
plt.plot(matrix_numpy_times, color="blue")
plt.plot(matrix_krylov_dim, matrix_krylov_tot_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()

Toto jsou skutečné naměřené časy, ale pro účely diskuse tyto křivky vyhladíme zprůměrováním přes několik sousedních bodů / dimenzí matice. To je provedeno níže:
smooth_numpy_times = []
smooth_krylov_times = []
# Choose the number of adjacent points over which to average forward;
# the same will be used backward.
smooth_steps = 10
# We will do this smoothing for all points/matrix dimensions
for i in range(len(matrix_krylov_tot_times)):
# Ensure we don't exceed the boundaries of our lists
start = max(0, i - smooth_steps)
end = min(len(matrix_krylov_tot_times) - 1, i + smooth_steps)
# Dummy variables for accumulating an average over adjacent points. This is done for both Krylov
# and the NumPy calculations.
smooth_count = 0
smooth_numpy_sum = 0
smooth_krylov_sum = 0
for j in range(start, end):
smooth_numpy_sum = smooth_numpy_sum + matrix_numpy_times[j]
smooth_krylov_sum = smooth_krylov_sum + matrix_krylov_tot_times[j]
smooth_count = smooth_count + 1
# Appending the averaged adjacent values to our new smooth lists
smooth_numpy_times.append(smooth_numpy_sum / smooth_count)
smooth_krylov_times.append(smooth_krylov_sum / smooth_count)
plt.plot(smooth_numpy_times, color="blue")
plt.plot(smooth_krylov_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (smoothed, microsec)") # Add y-axis label
plt.show()

Všimni si, že čas potřebný pro sestavení Krylovova podprostoru zpočátku překračuje čas potřebný pro úplnou diagonalizaci pomocí numpy. Jak ale roste velikost matice, Krylovova metoda se stává výhodnější. To platí i při snížení přijatelné chyby, avšak výhoda nastupuje při větší velikosti matice. Je to zajímavé a stojí za to to rozebrat.
Časová složitost numerické diagonalizace je (s určitými odchylkami v závislosti na algoritmu). Časová složitost generování ortonormální báze vektorů je také . Výhoda Krylovovy metody tedy není spojena s použitím ortonormální báze, ale s použitím konkrétní ortonormální báze, která efektivně vybírá vlastní hodnoty zájmu. Toto jsme již viděli z náčrtu důkazu v první části této lekce a je to klíčové pro záruky konvergence v Krylovových metodách. Shrňme, čeho jsme dosud dosáhli:
- Pro velmi velké matice může Krylovova metoda podprostorů poskytnout přibližné vlastní hodnoty v rámci požadovaných tolerancí rychleji než tradiční diagonalizační algoritmy.
- U takto velmi velkých matic je generování Krylovova podprostoru nejnáročnější částí metody Krylovova podprostoru.
- Efektivní způsob generování Krylovova podprostoru by tedy byl velmi cenný. A právě zde vstupuje do hry kvantový počítač.
Ověř si porozumění
Přečti si níže uvedenou otázku, zamysli se nad odpovědí a poté klikni na trojúhelník pro zobrazení řešení.
Viz vyhlazený graf časů diagonalizace v závislosti na dimenzi matice výše.
(a) Přibližně při jaké dimenzi matice se Krylovova metoda stala rychlejší podle tohoto grafu?
(b) Jaké aspekty výpočtu mohou změnit dimenzi, při níž se Krylovova metoda stane rychlejší?
Odpověď:
(a) Odpovědi se mohou lišit, pokud výpočet znovu spustíš, ale Krylovova metoda se stává rychlejší přibližně při dimenzi 80–85. (b) Možných odpovědí je mnoho. Mezi důležité faktory patří požadovaná přesnost a řídkost diagonalizovaných matic.
3. Krylov prostřednictvím časového vývoje
Vše, co jsme dosud popsali, lze provést klasicky. Jak a kdy bychom tedy použili kvantový počítač? Pro velmi velké matice může Krylovova metoda vyžadovat dlouhé výpočetní časy a velké množství paměti. Čas potřebný pro maticovou operaci na je v nejhorším případě . Dokonce i násobení řídkých matic vektorem (typický případ pro klasické Krylovovy řešiče) má časovou složitost . To se provádí pro každý vektor, který chceme mít v našem podprostoru. Dimenze podprostoru obvykle není výraznou frakcí a často se škáluje jako . Generování všech vektorů se tedy škáluje jako v nejhorším případě. Ačkoli existují i další kroky, jako je ortogonalizace, toto je dominantní škálování, které je třeba mít na paměti.
Kvantové výpočty nám umožňují změnit, které vlastnosti problému určují škálování potřebného času a zdrojů. Místo závislosti na velikosti matice napříč celým problémem uvidíme věci jako počet měření (shots) a počet nekomutujících Pauliho členů tvořících Hamiltonian. Pojďme prozkoumat, jak to funguje.
3.1 Časový vývoj
Vzpomeň si, že operátor, který provádí časový vývoj kvantového stavu, je (a v kvantových výpočtech se z notace velmi často vynechává). Jedním ze způsobů, jak takovouto exponenciální funkci operátoru pochopit a realizovat, je rozvinout ji do Taylorovy řady. Všimni si, že tato operace působící na nějaký počáteční vektor dává součet členů se stále vyššími mocninami aplikovanými na počáteční stav. Vypadá to, že Krylovův podprostor lze jednoduše vytvořit časovým vývojem počátečního odhadového stavu!
Háček spočívá v realizaci časového vývoje na skutečném kvantovém počítači. Mnohé členy Hamiltoniánu spolu nekomutují. Takže zatímco některé jednoduché exponenciální operátory, jako , odpovídají jednoduchým obvodům, obecné Hamiltoniány nikoli. A protože obsahují nekomutující členy, nemůžeme exponenciálu jednoduše rozložit na součin jednoduchých exponenciál, jak to lze s čísly.
To tedy není triviální, ale jde o dobře prostudovaný postup v kvantových výpočtech. Časový vývoj na kvantových počítačích provádíme pomocí procesu zvaného trotterizace, což je samo o sobě bohaté téma[10]. Na velmi vysoké úrovni však platí, že rozdělením časového vývoje na velmi malé kroky — řekněme kroků o délce — omezíme dopady nekomutativnosti členů.
kde .
Krylovův podprostor řádu r, který jsme v klasickém kontextu generovali přímo pomocí mocnin H, nazveme „mocninný Krylovův podprostor" (power Krylov subspace).
Nyní generujeme podobný prostor pomocí unitárního operátoru časového vývoje ; budeme ho označovat jako „unitární Krylovův prostor" . Mocninný Krylovův podprostor , který se používá klasicky, nelze přímo generovat na kvantovém počítači, protože není unitární operátor. Lze ukázat, že použití unitárního Krylovova podprostoru poskytuje podobné záruky konvergence jako mocninný Krylovův podprostor — konkrétně, chyba základního stavu konverguje efektivně za předpokladu, že počáteční stav má s pravým základním stavem překryv, který exponenciálně nevymizí, a za předpokladu, že mezi vlastními hodnotami existuje dostatečná mezera. Přesnější diskusi konvergence najdeš v Ref [1].
Zde se mocniny stávají různými časovými kroky (k-tá mocnina odpovídá posunu o čas ). Prvek podprostoru, jehož celkový čas časového vývoje je , označíme .
Náš Hamiltonian H můžeme promítnout do unitárního Krylovova podprostoru . Jinými slovy, vypočítáme každý maticový prvek v bázi . Tuto promítnutou matici budeme označovat .
3.2 Jak implementovat na kvantovém počítači
Maticové prvky jsou dány střednými hodnotami , které lze odhadnout pomocí kvantového počítače. Mej na paměti, že lze zapsat jako součet Pauli operátorů na různých qubitech a že ne všechny Pauli operátory lze měřit současně. Pauli členy lze roztřídit do skupin komutujících členů a všechny je najednou změřit. Může ale být zapotřebí mnoho takových skupin, aby pokryly všechny členy. Proto se stává důležitým počet různých komutujících skupin, na které lze členy rozdělit, .
Zde je Pauli řetězec tvaru nebo sada takových Pauli řetězců, které spolu navzájem komutují. Vzhledem k tomu, že lze zapsat jako takovýto součet měřitelných operátorů, lze následující výrazy pro maticové prvky realizovat pomocí primitiva Estimator Qiskit Runtime.
Kde jsou vektory unitárního Krylovova prostoru a jsou násobky zvoleného časového kroku . Na kvantovém počítači lze výpočet každého maticového prvku provést libovolným algoritmem, který nám umožňuje získat překryv kvantových stavů. V této lekci se zaměříme na Hadamardův test. Protože má dimenzi , bude mít Hamiltonian promítnutý do podprostoru rozměry . Pokud je dostatečně malé (obecně stačí pro dosažení konvergence odhadů vlastních hodnot), lze promítnutý Hamiltonian snadno diagonalizovat klasicky. Avšak nelze přímo diagonalizovat kvůli neortogonalitě vektorů Krylovova prostoru. Budeme muset změřit jejich překryvy a sestavit matici .
To nám umožňuje řešit problém vlastních hodnot v neortogonálním prostoru (nazývaný také zobecněný problém vlastních hodnot):
Z řešení tohoto zobecněného problému vlastních hodnot lze poté získat odhady vlastních hodnot a vlastních stavů . Odhad energie základního stavu se například získá jako nejmenší vlastní hodnota a základní stav z odpovídajícího vlastního vektoru . Koeficienty v určují příspěvek různých vektorů, které rozpínají .
Zobecněný problém vlastních hodnot
Proč nemůžeme jednoduše diagonalizovat ? Protože obsahuje informaci o geometrii Krylovovy báze (která je neortogonální ve všech případech kromě velmi speciálních), samotné nepopisuje projekci celého Hamiltoniánu, a jeho vlastní hodnoty tedy nemají žádný zvláštní vztah k vlastním hodnotám celého Hamiltoniánu — mohou být libovolné náhodné hodnoty. K získání přibližných vlastních hodnot a vlastních vektorů odpovídajících projekci celého Hamiltoniánu do Krylovova prostoru je nutné řešit zobecněný problém vlastních hodnot.
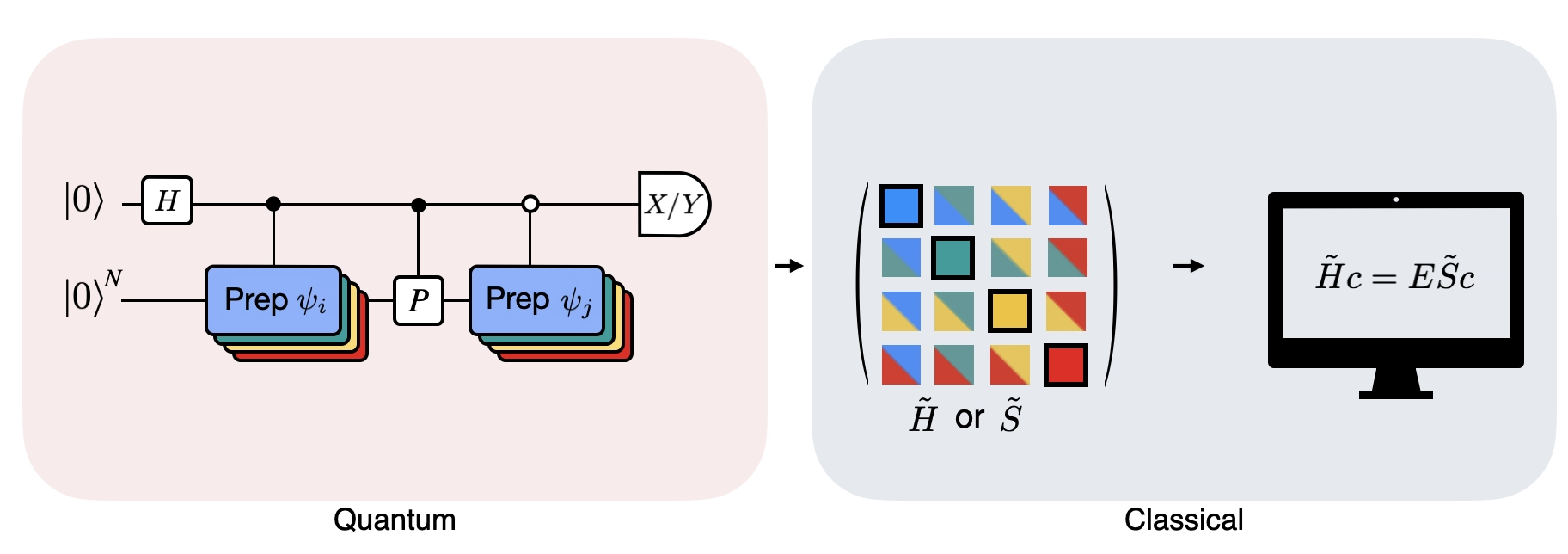
Obrázek ukazuje obvodovou reprezentaci modifikovaného Hadamardova testu, metody používané k výpočtu překryvu mezi různými kvantovými stavy. Pro každý maticový prvek se provede Hadamardův test mezi stavy a . To je v obrázku zvýrazněno barevným schématem pro maticové prvky a odpovídající operace , . K výpočtu všech maticových prvků promítnutého Hamiltoniánu je tedy potřeba sada Hadamardových testů pro všechny možné kombinace vektorů Krylovova prostoru. Horní vodič v obvodu Hadamardova testu je pomocný qubit (ancilla), který se měří buď v bázi X nebo Y; jeho střední hodnota určuje hodnotu překryvu mezi stavy. Dolní vodič představuje všechny qubity systémového Hamiltoniánu. Operace připraví systémový qubit ve stavu řízeném stavem pomocného qubitu (stejně tak pro ) a operace představuje Pauli rozklad systémového Hamiltoniánu . Implementace tohoto postupu na kvantovém počítači je podrobněji ukázána níže.
4. Krylov kvantová diagonalizace na kvantovém počítači
Nyní implementujeme Krylov kvantovou diagonalizaci na skutečném kvantovém počítači. Začněme importem některých užitečných balíčků.
import numpy as np
import scipy as sp
import matplotlib.pylab as plt
from typing import Union, List
import warnings
from qiskit.quantum_info import SparsePauliOp, Pauli
from qiskit.circuit import Parameter
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit.library import PauliEvolutionGate
from qiskit.synthesis import LieTrotter
# from qiskit.providers.fake_provider import Fake20QV1
from qiskit_ibm_runtime import QiskitRuntimeService, EstimatorV2 as Estimator, Batch
import itertools as it
warnings.filterwarnings("ignore")
Níže definujeme funkci pro řešení zobecněného problému vlastních hodnot, který jsme právě popsali výše.
def solve_regularized_gen_eig(
h: np.ndarray,
s: np.ndarray,
threshold: float,
k: int = 1,
return_dimn: bool = False,
) -> Union[float, List[float]]:
"""
Method for solving the generalized eigenvalue problem with regularization
Args:
h (numpy.ndarray):
The effective representation of the matrix in our Krylov subspace
s (numpy.ndarray):
The matrix of overlaps between vectors of our Krylov subspace
threshold (float):
Cut-off value for the eigenvalue of s
k (int):
Number of eigenvalues to return
return_dimn (bool):
Whether to return the size of the regularized subspace
Returns:
lowest k-eigenvalue(s) that are the solution of the regularized generalized eigenvalue problem
"""
s_vals, s_vecs = sp.linalg.eigh(s)
s_vecs = s_vecs.T
good_vecs = np.array([vec for val, vec in zip(s_vals, s_vecs) if val > threshold])
h_reg = good_vecs.conj() @ h @ good_vecs.T
s_reg = good_vecs.conj() @ s @ good_vecs.T
if k == 1:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][0], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][0]
else:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][:k], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][:k]
Přinejmenším při počátečním benchmarkingu je užitečné znát přesné klasické řešení pro ověření chování konvergence. Níže uvedená funkce vypočítá energii základního stavu Hamiltoniánu; jako argumenty přijímá Hamiltonian a počet qubitů.
def single_particle_gs(H_op, n_qubits):
"""
Find the ground state of the single particle(excitation) sector
"""
H_x = []
for p, coeff in H_op.to_list():
H_x.append(set([i for i, v in enumerate(Pauli(p).x) if v]))
H_z = []
for p, coeff in H_op.to_list():
H_z.append(set([i for i, v in enumerate(Pauli(p).z) if v]))
H_c = H_op.coeffs
print("n_sys_qubits", n_qubits)
n_exc = 1
sub_dimn = int(sp.special.comb(n_qubits + 1, n_exc))
print("n_exc", n_exc, ", subspace dimension", sub_dimn)
few_particle_H = np.zeros((sub_dimn, sub_dimn), dtype=complex)
sparse_vecs = [
set(vec) for vec in it.combinations(range(n_qubits + 1), r=n_exc)
] # list all of the possible sets of n_exc indices of 1s in n_exc-particle states
m = 0
for i, i_set in enumerate(sparse_vecs):
for j, j_set in enumerate(sparse_vecs):
m += 1
if len(i_set.symmetric_difference(j_set)) <= 2:
for p_x, p_z, coeff in zip(H_x, H_z, H_c):
if i_set.symmetric_difference(j_set) == p_x:
sgn = ((-1j) ** len(p_x.intersection(p_z))) * (
(-1) ** len(i_set.intersection(p_z))
)
else:
sgn = 0
few_particle_H[i, j] += sgn * coeff
gs_en = min(np.linalg.eigvalsh(few_particle_H))
print("single particle ground state energy: ", gs_en)
return gs_en
4.1 Krok 1: Namapuj problém na kvantové obvody a operátory
Nyní definujeme Hamiltonian. Ten se liší od výše uvedené funkce v tom, že zmíněná funkce bere Hamiltonian jako argument a vrací pouze základní stav – a dělá to klasicky. Hamiltonian, který zde definujeme, určuje energetické hladiny všech energetických vlastních stavů a lze ho sestavit pomocí Pauliho operátorů a implementovat na kvantovém počítači.
Volíme Hamiltonian odpovídající řetězci spinů, které mohou mít libovolnou orientaci v prostoru – tzv. „Heisenbergův řetězec". Předpokládáme, že spin může být ovlivněn svými nejbližšími sousedy (spiny a ), ale ne vzdálenějšími sousedy. Zároveň připouštíme, že interakce mezi spiny může být různá v závislosti na tom, podél které osy spiny míří. Tato asymetrie někdy vzniká například kvůli struktuře krystalové mřížky, do níž jsou spiny zasazeny.
# Define problem Hamiltonian.
n_qubits = 10
# coupling strength for XX, YY, and ZZ interactions
JX = 1
JY = 3
JZ = 2
# Define the Hamiltonian:
H_int = [["I"] * n_qubits for _ in range(3 * (n_qubits - 1))]
for i in range(n_qubits - 1):
H_int[i][i] = "Z"
H_int[i][i + 1] = "Z"
for i in range(n_qubits - 1):
H_int[n_qubits - 1 + i][i] = "X"
H_int[n_qubits - 1 + i][i + 1] = "X"
for i in range(n_qubits - 1):
H_int[2 * (n_qubits - 1) + i][i] = "Y"
H_int[2 * (n_qubits - 1) + i][i + 1] = "Y"
H_int = ["".join(term) for term in H_int]
H_tot = [
(term, JZ)
if term.count("Z") == 2
else (term, JY)
if term.count("Y") == 2
else (term, JX)
for term in H_int
]
# Get operator
H_op = SparsePauliOp.from_list(H_tot)
print(H_tot)
[('ZZIIIIIIII', 2), ('IZZIIIIIII', 2), ('IIZZIIIIII', 2), ('IIIZZIIIII', 2), ('IIIIZZIIII', 2), ('IIIIIZZIII', 2), ('IIIIIIZZII', 2), ('IIIIIIIZZI', 2), ('IIIIIIIIZZ', 2), ('XXIIIIIIII', 1), ('IXXIIIIIII', 1), ('IIXXIIIIII', 1), ('IIIXXIIIII', 1), ('IIIIXXIIII', 1), ('IIIIIXXIII', 1), ('IIIIIIXXII', 1), ('IIIIIIIXXI', 1), ('IIIIIIIIXX', 1), ('YYIIIIIIII', 3), ('IYYIIIIIII', 3), ('IIYYIIIIII', 3), ('IIIYYIIIII', 3), ('IIIIYYIIII', 3), ('IIIIIYYIII', 3), ('IIIIIIYYII', 3), ('IIIIIIIYYI', 3), ('IIIIIIIIYY', 3)]
Níže uvedený kód omezí Hamiltonian na jednočásticové stavy a pomocí spektrální normy stanoví vhodnou velikost časového kroku . Heuristicky volíme hodnotu časového kroku dt (na základě horních mezí normy Hamiltonianu). Odkaz [9] ukázal, že dostatečně malý časový krok je , a že je výhodné tuto hodnotu spíše podceňovat než přeceňovat – přecenění totiž může umožnit, aby příspěvky z vysokoenergetických stavů znehodnotily i optimální stav v Krylovově prostoru. Na druhou stranu příliš malé vede k horšímu podmínění Krylovova podprostoru, protože bázové vektory Krylovovy báze se pak od časového kroku k časovému kroku méně liší.
# Get Hamiltonian restricted to single-particle states
single_particle_H = np.zeros((n_qubits, n_qubits))
for i in range(n_qubits):
for j in range(i + 1):
for p, coeff in H_op.to_list():
p_x = Pauli(p).x
p_z = Pauli(p).z
if all(p_x[k] == ((i == k) + (j == k)) % 2 for k in range(n_qubits)):
sgn = ((-1j) ** sum(p_z[k] and p_x[k] for k in range(n_qubits))) * (
(-1) ** p_z[i]
)
else:
sgn = 0
single_particle_H[i, j] += sgn * coeff
for i in range(n_qubits):
for j in range(i + 1, n_qubits):
single_particle_H[i, j] = np.conj(single_particle_H[j, i])
# Set dt according to spectral norm
dt = np.pi / np.linalg.norm(single_particle_H, ord=2)
dt
np.float64(0.17453292519943295)
Určeme maximální Krylovovu dimenzi, kterou chceme použít, přičemž budeme kontrolovat konvergenci i při menších dimenzích. Také určíme počet Trotterových kroků použitých při časovém vývoji. Pro účely této lekce zvolíme malou Krylovovu dimenzi pouhých 4. To je v kontextu realistických aplikací velmi omezené, ale pro tento příklad to postačí. V pozdějších lekcích prozkoumáme metody, které nám umožní škálovat a promítat naše Hamiltoniany na větší podprostory.
# Set parameters for quantum Krylov algorithm
krylov_dim = 4 # size of krylov subspace
num_trotter_steps = 4
dt_circ = dt / num_trotter_steps
Příprava stavu
Zvol referenční stav , který má nenulový překryv se základním stavem. Pro tento Hamiltonian použijeme jako referenční stav stav s excitací ve středním qubitu .
qc_state_prep = QuantumCircuit(n_qubits)
qc_state_prep.x(int(n_qubits / 2) + 1)
qc_state_prep.draw("mpl", scale=0.5)

Časový vývoj
Operátor časového vývoje generovaný daným Hamiltonianem lze realizovat pomocí Lie-Trotterovy aproximace. Pro jednoduchost použijeme v obvodu časového vývoje vestavěný PauliEvolutionGate. Obecná syntaxe je tato.
t = Parameter("t")
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=t, synthesis=LieTrotter(reps=num_trotter_steps)
)
qr = QuantumRegister(n_qubits)
qc_evol = QuantumCircuit(qr)
qc_evol.append(evol_gate, qargs=qr)
<qiskit.circuit.instructionset.InstructionSet at 0x7ccaa4664250>
Níže v Hadamardově testu použijeme verzi tohoto obvodu, která provádí kroky vpřed o čas .
Hadamard test
Připomeňme, že chceme vypočítat maticové prvky jak , tak Gramovy matice pomocí Hadamardova testu. Pojďme si zopakovat, jak to funguje v tomto kontextu, a zaměřme se nejprve na sestrojení Celý postup je graficky znázorněn níže. Vrstvy barevných bloků přípravy stavů připomínají, že tento postup se provádí pro všechny kombinace a v našem podprostoru.
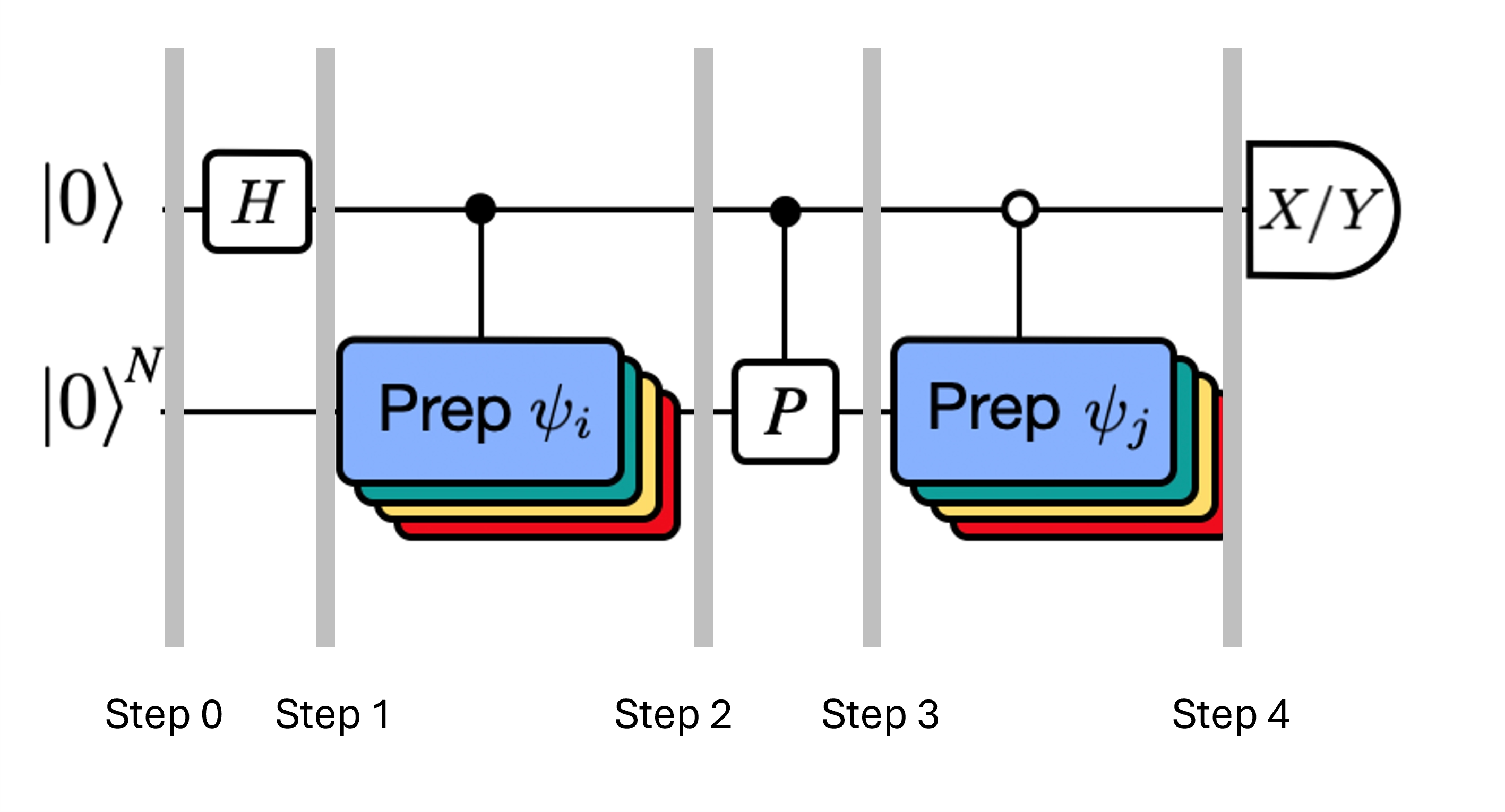
Stavy systému v označených krocích jsou:
Zde je Pauliho člen v rozkladu Hamiltoniánu (všimni si, že nemůže jít o lineární kombinaci více komutujících Pauliho členů, protože ta by nebyla unitární -- seskupování je možné pomocí jiné konstrukce, kterou ukážeme později). , jsou řízené operace, které připravují vektory , unitárního Krylovova prostoru, přičemž . Měření a aplikovaná na tento obvod vypočítají reálnou, respektive imaginární část požadovaných maticových prvků.
Počínaje krokem 4 výše, aplikuj Hadamardovo hradlo na nultý qubit.
Poté změř buď , nebo .
Z identity . Obdobně měření dává
Přidáme tyto kroky k časové evoluci, kterou jsme nastavili dříve, a zapíšeme následující.
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
## Create the time-evo op dagger circuit
evol_gate_d = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
evol_gate_d = evol_gate_d.inverse()
# Put pieces together
qc_reg = QuantumRegister(n_qubits)
qc_temp = QuantumCircuit(qc_reg)
qc_temp.compose(qc_state_prep, inplace=True)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate, qargs=qc_reg)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate_d, qargs=qc_reg)
qc_temp.compose(qc_state_prep.inverse(), inplace=True)
# Create controlled version of the circuit
controlled_U = qc_temp.to_gate().control(1)
# Create hadamard test circuit for real part
qr = QuantumRegister(n_qubits + 1)
qc_real = QuantumCircuit(qr)
qc_real.h(0)
qc_real.append(controlled_U, list(range(n_qubits + 1)))
qc_real.h(0)
print("Circuit for calculating the real part of the overlap in S via Hadamard test")
qc_real.draw("mpl", fold=-1, scale=0.5)
Circuit for calculating the real part of the overlap in S via Hadamard test

Již jsme varovali před hloubkou, kterou mají Trotterovy obvody. Provádění Hadamardova testu za těchto podmínek může vést k ještě hlubšímu obvodu, zejména po rozkladu na nativní hradla. Tato hloubka se ještě zvýší, pokud zohledníme topologii zařízení. Proto je dobré před využitím jakéhokoliv času na kvantovém počítači zkontrolovat hloubku dvouqubitových operací našeho obvodu.
print(
"Number of layers of 2Q operations",
qc_real.decompose(reps=2).depth(lambda x: x[0].num_qubits == 2),
)
Number of layers of 2Q operations 14401
Obvod takové hloubky nemůže na moderních kvantových počítačích vrátit použitelné výsledky. Pokud chceme sestavit a potřebujeme lepší přístup. To je důvod pro zavedení efektivního Hadamardova testu, který je popsán níže.
4. 2 Krok 2. Optimalizace obvodů a operátorů pro cílový hardware
Efektivní Hadamard test
Hluboké obvody pro Hadamardův test, které jsme získali, lze optimalizovat zavedením některých aproximací a opřením se o určité předpoklady o modelovém Hamiltoniánu. Uvažuj například následující obvod pro Hadamardův test:
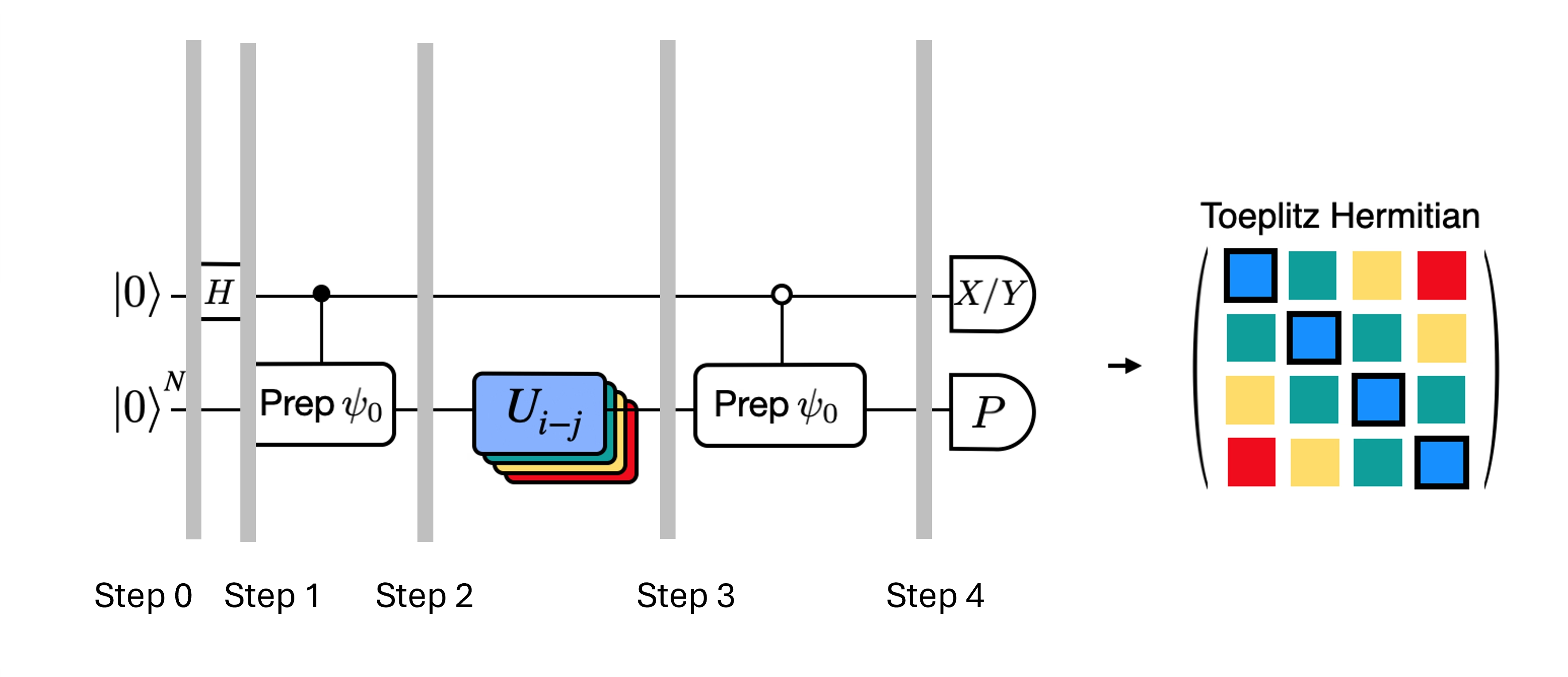
Předpokládejme, že dokážeme klasicky vypočítat , vlastní hodnotu pod Hamiltoniánem . Tato podmínka je splněna, pokud Hamiltonián zachovává symetrii U(1). Ačkoli se to může zdát jako silný předpoklad, existuje mnoho případů, kdy je bezpečné předpokládat, že existuje vakuový stav (v tomto případě se zobrazuje na stav ), který není ovlivněn působením Hamiltoniánu. To platí například pro chemické Hamiltoniány popisující stabilní molekuly (kde je zachován počet elektronů). Vzhledem k tomu, že hradlo připravuje požadovaný referenční stav , například pro přípravu HF stavu pro chemii je součinem jednoqubitových hradel NOT, takže řízená- je jen součinem hradel CNOT. Výše uvedený obvod pak před měřením implementuje následující stav:
kde jsme použili klasicky simulovatelný fázový posun z kroku 2 na krok 3. Střední hodnoty jsou tedy
Díky těmto předpokladům jsme dokázali vyjádřit střední hodnoty požadovaných operátorů s menším počtem řízených operací. Ve skutečnosti potřebujeme implementovat pouze řízenou přípravu stavu , nikoli řízené časové evoluce. Přeformulování výpočtu výše nám umožní výrazně snížit hloubku výsledných obvodů.
Poznamenejme, že jako bonus -- protože Pauliho operátor se nyní objevuje jako měření na konci obvodu namísto jako řízené hradlo uprostřed -- lze jej měřit společně s dalšími komutujícími Pauliho operátory, jako v rozkladu uvedeném výše.
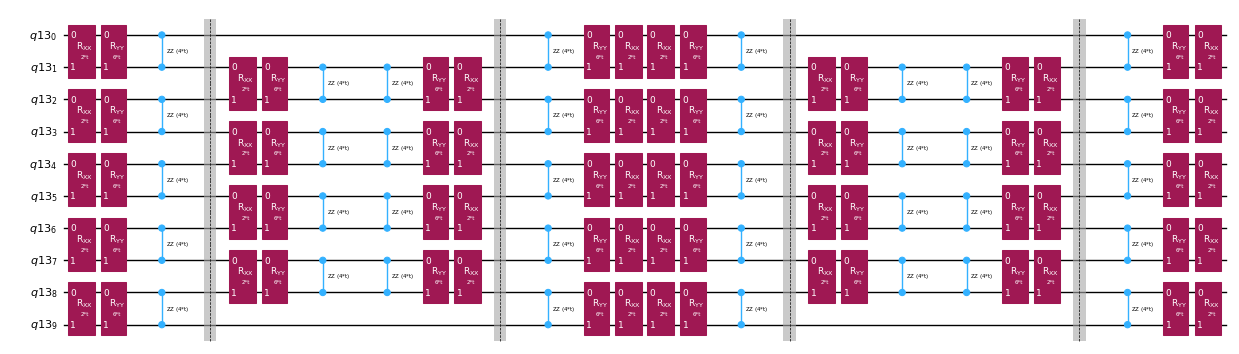
Rozložení operátoru časového vývoje pomocí Trotter dekompozice
Místo přesné implementace operátoru časového vývoje můžeme použít Trotter dekompozici a implementovat jeho aproximaci. Opakováním Trotter dekompozice určitého řádu vícekrát dosáhneme dalšího snížení chyby vzniklé z aproximace. V následujícím textu přímo sestavíme Trotter implementaci nejefektivnějším způsobem pro interakční graf Hamiltoniánu, který uvažujeme (pouze interakce nejbližších sousedů). V praxi vkládáme Pauli rotace , , s vazebními konstantami , , a parametrizovaným úhlem , které odpovídají přibližné implementaci . Vzhledem k rozdílu v definici Pauli rotací a časového vývoje, který se snažíme implementovat, budeme muset použít parametr pro dosažení časového vývoje . Navíc obrátíme pořadí operací pro lichý počet opakování Trotter kroků, což je funkčně ekvivalentní, ale umožňuje syntézu sousedních operací do jediného unitárního operátoru. To dává mnohem mělčí obvod než ten, který se získá pomocí generické funkcionality PauliEvolutionGate().
t = Parameter("t")
# Create instruction for rotation about XX+YY-ZZ:
Rxyz_circ = QuantumCircuit(2)
Rxyz_circ.rxx(2 * JX * t, 0, 1)
Rxyz_circ.ryy(2 * JY * t, 0, 1)
Rxyz_circ.rzz(2 * JZ * t, 0, 1)
Rxyz_instr = Rxyz_circ.to_instruction(label="R J_x XX + J_y YY + J_z ZZ")
interaction_list = [
[[i, i + 1] for i in range(0, n_qubits - 1, 2)],
[[i, i + 1] for i in range(1, n_qubits - 1, 2)],
] # linear chain
qr = QuantumRegister(n_qubits)
trotter_step_circ = QuantumCircuit(qr)
for i, color in enumerate(interaction_list):
for interaction in color:
trotter_step_circ.append(Rxyz_instr, interaction)
if i < len(interaction_list) - 1:
trotter_step_circ.barrier()
reverse_trotter_step_circ = trotter_step_circ.reverse_ops()
qc_evol = QuantumCircuit(qr)
for step in range(num_trotter_steps):
if step % 2 == 0:
qc_evol = qc_evol.compose(trotter_step_circ)
else:
qc_evol = qc_evol.compose(reverse_trotter_step_circ)
qc_evol.decompose().draw("mpl", fold=-1, scale=0.5)
Pro tento efektivní Hadamardův test opět připravíme počáteční stav.
control = 0
excitation = int(n_qubits / 2) + 1
controlled_state_prep = QuantumCircuit(n_qubits + 1)
controlled_state_prep.cx(control, excitation)
controlled_state_prep.draw("mpl", fold=-1, scale=0.5)

Šablonové obvody pro výpočet maticových prvků a pomocí Hadamardova testu
Jediný rozdíl mezi obvody použitými v Hadamardově testu bude fáze v operátoru časového vývoje a měřené observabely. Proto můžeme připravit šablonový obvod, který reprezentuje obecný obvod pro Hadamardův test, se zástupnými místy pro hradla závisející na operátoru časového vývoje.
# Parameters for the template circuits
parameters = []
for idx in range(1, krylov_dim):
parameters.append(dt_circ * (idx))
# Create modified hadamard test circuit
qr = QuantumRegister(n_qubits + 1)
qc = QuantumCircuit(qr)
qc.h(0)
qc.compose(controlled_state_prep, list(range(n_qubits + 1)), inplace=True)
qc.barrier()
qc.compose(qc_evol, list(range(1, n_qubits + 1)), inplace=True)
qc.barrier()
qc.x(0)
qc.compose(controlled_state_prep.inverse(), list(range(n_qubits + 1)), inplace=True)
qc.x(0)
qc.decompose().draw("mpl", fold=-1)
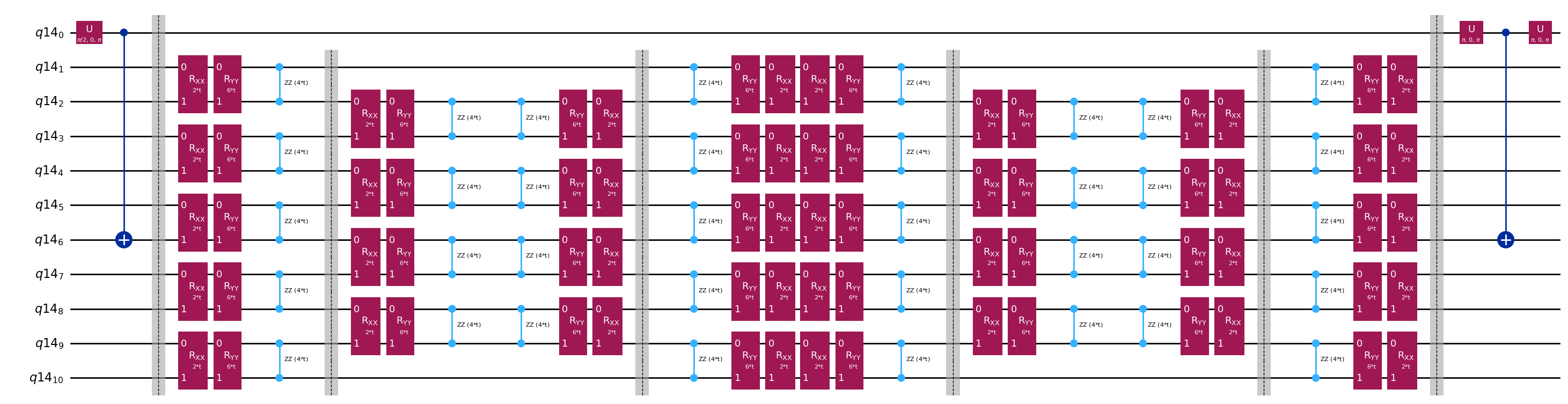
print(
"The optimized circuit has 2Q gates depth: ",
qc.decompose().decompose().depth(lambda x: x[0].num_qubits == 2),
)
The optimized circuit has 2Q gates depth: 50
Tato hloubka je výrazně snížena ve srovnání s původním Hadamardovým testem. Tato hloubka je zvládnutelná na moderních kvantových počítačích, ačkoli je stále poměrně vysoká. Budeme muset použít nejmodernější potlačení chyb, abychom získali užitečné výsledky.
Vyber backend, na kterém spustíš kvantový Krylov výpočet, abychom mohli náš obvod transpilovat pro spuštění na tom kvantovém počítači.
# Use the least-busy backend or specify a quantum computer using the syntax commented out below.
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
# Or you may choose a specify backend and channel if necessary for your workflow.
# service = QiskitRuntimeService(channel="ibm_quantum_platform")
# backend = service.backend("ibm_fez")
Nyní transpilujeme naše obvody a operátory.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
basis_gates = list(target.operation_names)
pm = generate_preset_pass_manager(
optimization_level=3, backend=backend, basis_gates=basis_gates
)
qc_trans = pm.run(qc)
print(qc_trans.depth(lambda x: x[0].num_qubits == 2))
print(qc_trans.count_ops())
qc_trans.draw("mpl", fold=-1, idle_wires=False, scale=0.5)
36
OrderedDict([('rz', 410), ('sx', 361), ('cz', 156), ('x', 18), ('barrier', 6)])

Po optimalizaci je hloubka dvouqubitových hradel našeho transpilovaného obvodu dále snížena.
4.3 Krok 3. Spuštění pomocí primitivu Qiskit Runtime
Nyní vytvoříme PUBy pro spuštění s Estimatorem.
# Define observables to measure for S
observable_S_real = "I" * (n_qubits) + "X"
observable_S_imag = "I" * (n_qubits) + "Y"
observable_op_real = SparsePauliOp(
observable_S_real
) # define a sparse pauli operator for the observable
observable_op_imag = SparsePauliOp(observable_S_imag)
layout = qc_trans.layout # get layout of transpiled circuit
observable_op_real = observable_op_real.apply_layout(
layout
) # apply physical layout to the observable
observable_op_imag = observable_op_imag.apply_layout(layout)
observable_S_real = (
observable_op_real.paulis.to_labels()
) # get the label of the physical observable
observable_S_imag = observable_op_imag.paulis.to_labels()
observables_S = [[observable_S_real], [observable_S_imag]]
# Define observables to measure for H
# Hamiltonian terms to measure
observable_list = []
for pauli, coeff in zip(H_op.paulis, H_op.coeffs):
# print(pauli)
observable_H_real = pauli[::-1].to_label() + "X"
observable_H_imag = pauli[::-1].to_label() + "Y"
observable_list.append([observable_H_real])
observable_list.append([observable_H_imag])
layout = qc_trans.layout
observable_trans_list = []
for observable in observable_list:
observable_op = SparsePauliOp(observable)
observable_op = observable_op.apply_layout(layout)
observable_trans_list.append([observable_op.paulis.to_labels()])
observables_H = observable_trans_list
# Define a sweep over parameter values
params = np.vstack(parameters).T
# Estimate the expectation value for all combinations of
# observables and parameter values, where the pub result will have
# shape (# observables, # parameter values).
pub = (qc_trans, observables_S + observables_H, params)
Obvody pro lze spočítat klasicky. Provedeme tento výpočet dříve, než přejdeme k případu s využitím kvantového počítače.
from qiskit.quantum_info import StabilizerState, Pauli
qc_cliff = qc.assign_parameters({t: 0})
# Get expectation values from experiment
S_expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "X")
)
S_expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "Y")
)
# Get expectation values
S_expval = S_expval_real + 1j * S_expval_imag
H_expval = 0
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Get expectation values from experiment
expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "X")
)
expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "Y")
)
expval = expval_real + 1j * expval_imag
# Fill-in matrix elements
H_expval += coeff * expval
print(H_expval)
(10+0j)
Přestože se nám podařilo výrazně snížit hloubku hradel pomocí efektivního Hadamardova testu, je hloubka stále dostatečně velká, aby vyžadovala nejmodernější korekci chyb. Níže uvádíme vlastnosti používané korekce. Všechny použité metody jsou důležité, ale zvláštní zmínku si zaslouží pravděpodobnostní amplifikace chyb (PEA). Tato mocná technika je spojena se značnou kvantovou režií. Výpočet zde provedený může na skutečném kvantovém počítači trvat 20 minut i více. Možná si budeš chtít pohrát s níže uvedenými parametry a zvýšit nebo snížit přesnost, a tím i režii. Výchozí nastavení níže poskytuje výsledky s vysokou věrností.
# Experiment options
num_randomizations = 300
num_randomizations_learning = 20
max_batch_circuits = 20
shots_per_randomization = 100
learning_pair_depths = [0, 4, 24]
noise_factors = [1, 1.3, 1.6]
# Base option formatting
options = {
# Builtin resilience settings for ZNE
"resilience": {
"measure_mitigation": True,
"zne_mitigation": True,
"zne": {"noise_factors": noise_factors},
# TREX noise learning configuration
"measure_noise_learning": {
"num_randomizations": num_randomizations_learning,
"shots_per_randomization": shots_per_randomization,
},
# PEA noise model configuration
"layer_noise_learning": {
"max_layers_to_learn": 10,
"layer_pair_depths": learning_pair_depths,
"shots_per_randomization": shots_per_randomization,
"num_randomizations": num_randomizations_learning,
},
},
# Randomization configuration
"twirling": {
"num_randomizations": num_randomizations,
"shots_per_randomization": shots_per_randomization,
"strategy": "all",
},
# Experimental settings for PEA method
"experimental": {
# # Just in case, disable any further qiskit transpilation not related to twirling / DD
# "skip_transpilation": True,
# Execution configuration
"execution": {
"max_pubs_per_batch_job": max_batch_circuits,
"fast_parametric_update": True,
},
# Error Mitigation configuration
"resilience": {
# ZNE Configuration
"zne": {
"amplifier": "pea",
"return_all_extrapolated": True,
"return_unextrapolated": True,
"extrapolated_noise_factors": [0] + noise_factors,
}
},
},
}
Nakonec spustíme obvody pro a pomocí Estimatoru.
# This job required 17 minutes of QPU time to run on a Heron r2 processor. This is only an estimate.
# Your execution time may vary.
with Batch(backend=backend) as batch:
# Estimator
estimator = Estimator(mode=batch, options=options)
job = estimator.run([pub], precision=1)
4.4 Krok 4. Následné zpracování a analýza výsledků
Z kvantového počítače jsme získali jednotlivé prvky matic a komutující skupiny Pauliho operátorů, které tvoří prvky matice . Tyto členy je třeba zkombinovat, abychom mohli obnovit naše matice a vyřešit zobecněný problém vlastních čísel.
# Store the outputs as 'results'.
results = job.result()[0]
Výpočet efektivního Hamiltonianu a matic překryvu
Nejprve vypočítej fázi, kterou stav akumuluje během neřízené časové evoluce
prefactors = [
np.exp(-1j * sum([c for p, c in H_op.to_list() if "Z" in p]) * i * dt)
for i in range(1, krylov_dim)
]
Jakmile máme výsledky spuštění obvodů, můžeme data post-procesovat a vypočítat maticové elementy
# Assemble S, the overlap matrix of dimension D:
S_first_row = np.zeros(krylov_dim, dtype=complex)
S_first_row[0] = 1 + 0j
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[0][0][i] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[1][0][i] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
S_first_row[i + 1] += prefactors[i] * expval
S_first_row_list = S_first_row.tolist() # for saving purposes
S_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in it.product(range(krylov_dim), repeat=2):
if i >= j:
S_circ[j, i] = S_first_row[i - j]
else:
S_circ[j, i] = np.conj(S_first_row[j - i])
from sympy import Matrix
Matrix(S_circ)
A maticové elementy
import itertools
# Assemble S, the overlap matrix of dimension D:
H_first_row = np.zeros(krylov_dim, dtype=complex)
H_first_row[0] = H_expval
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[2 + 2 * obs_idx][0][
i
] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[2 + 2 * obs_idx + 1][0][
i
] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
H_first_row[i + 1] += prefactors[i] * coeff * expval
H_first_row_list = H_first_row.tolist()
H_eff_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in itertools.product(range(krylov_dim), repeat=2):
if i >= j:
H_eff_circ[j, i] = H_first_row[i - j]
else:
H_eff_circ[j, i] = np.conj(H_first_row[j - i])
from sympy import Matrix
Matrix(H_eff_circ)
Nakonec můžeme vyřešit zobecněný problém vlastních čísel pro :
a získat odhad energie základního stavu
gnd_en_circ_est_list = []
for d in range(1, krylov_dim + 1):
# Solve generalized eigenvalue problem
gnd_en_circ_est = solve_regularized_gen_eig(
H_eff_circ[:d, :d], S_circ[:d, :d], threshold=1e-1
)
gnd_en_circ_est_list.append(gnd_en_circ_est)
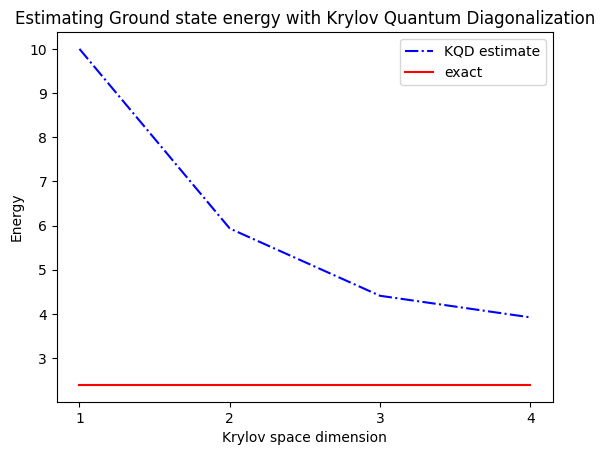
print("The estimated ground state energy is: ", gnd_en_circ_est)
The estimated ground state energy is: 10.0
The estimated ground state energy is: 5.933953916292923
The estimated ground state energy is: 4.4101773995740645
The estimated ground state energy is: 3.921288588521255
Pro sektor s jednou částicí můžeme efektivně spočítat základní stav tohoto sektoru Hamiltonianu klasicky
gs_en = single_particle_gs(H_op, n_qubits)
n_sys_qubits 10
n_exc 1 , subspace dimension 11
single particle ground state energy: 2.391547869638771
len(H_op)
27
plt.plot(
range(1, krylov_dim + 1),
gnd_en_circ_est_list,
color="blue",
linestyle="-.",
label="KQD estimate",
)
plt.plot(
range(1, krylov_dim + 1),
[gs_en] * krylov_dim,
color="red",
linestyle="-",
label="exact",
)
plt.xticks(range(1, krylov_dim + 1), range(1, krylov_dim + 1))
plt.legend()
plt.xlabel("Krylov space dimension")
plt.ylabel("Energy")
plt.title("Estimating Ground state energy with Krylov Quantum Diagonalization")
plt.show()
5. Diskuze a rozšíření
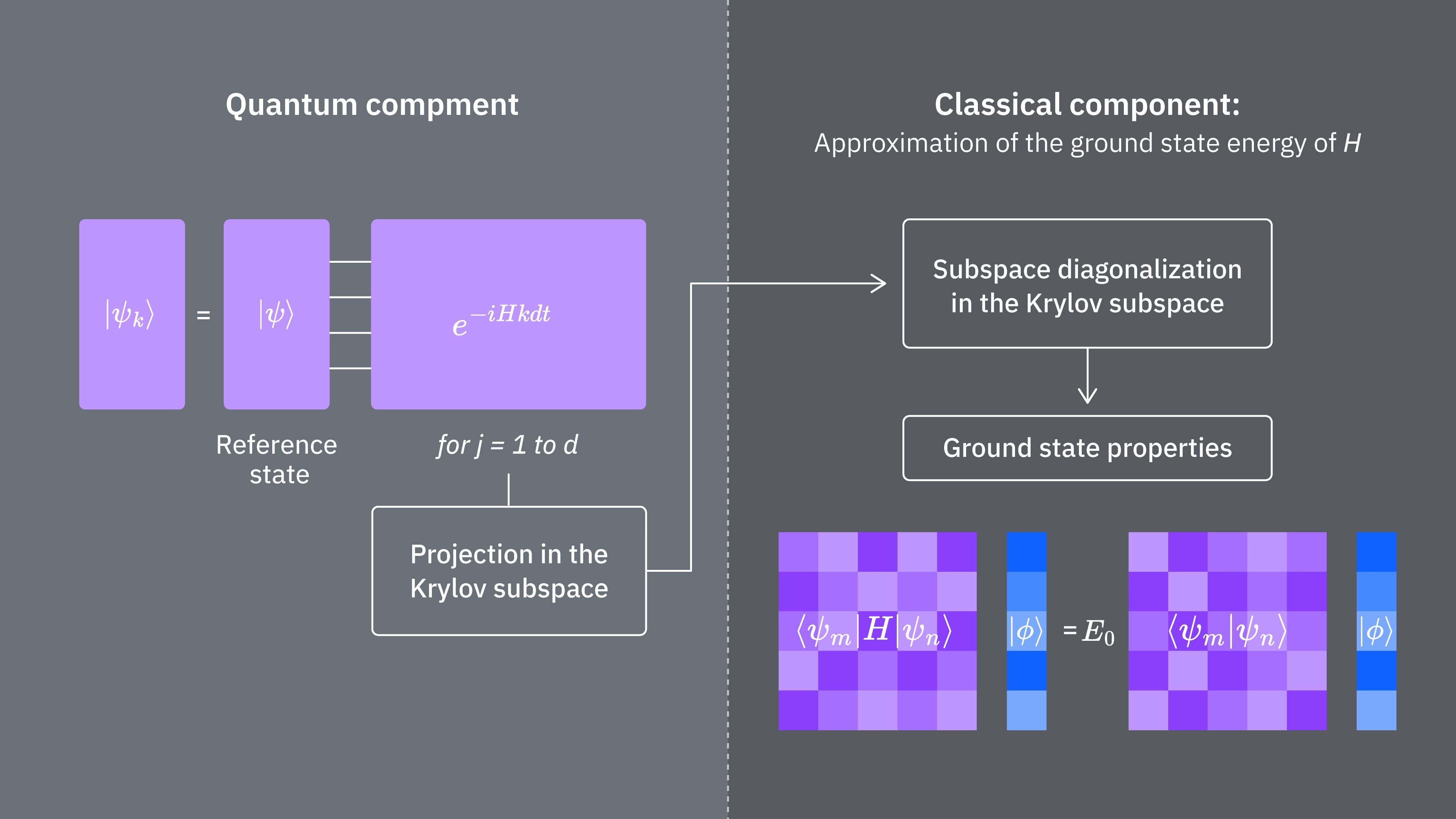
Shrňme to: začínáme s referenčním stavem, který pak vyvíjíme po různě dlouhé časové úseky, abychom vygenerovali unitární Krylovský podprostor. Náš Hamiltonian promítneme na tento podprostor. Odhadneme také překryvy vektorů podprostoru. Nakonec klasicky vyřešíme nízkodimenzionální zobecněný problém vlastních čísel.
Porovnejme, co určuje výpočetní náklady při použití Krylovské techniky klasicky a kvantově. Mezi klasickým a kvantovým přístupem neexistují dokonalé analogie pro všechny kroky. Tato tabulka shrnuje škálování různých kroků pro srovnání.
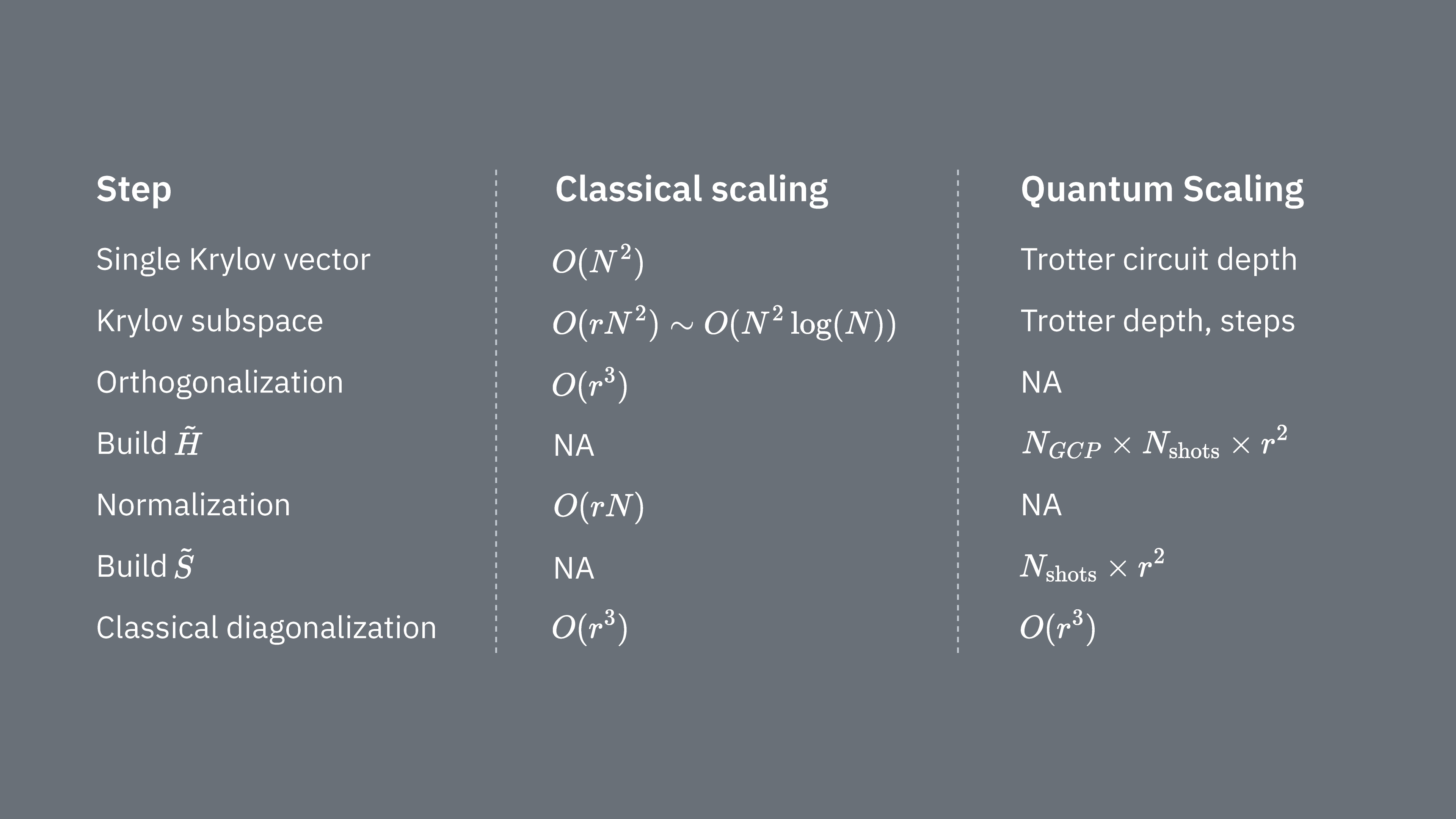
Připomeňme, že Hamiloniany obecně obsahují členy, které nelze měřit současně (protože spolu navzájem nekomutují). Členy Hamiltonianu třídíme do skupin komutujících Pauliho operátorů, které lze všechny měřit najednou, a k pokrytí všech nekomutujících členů může být potřeba mnoho takových skupin. Sestavení na kvantovém počítači vyžaduje samostatná měření pro každou skupinu komutujících Pauliho řetězců v Hamiltonianu, přičemž každá taková skupina vyžaduje mnoho shotů. Musíme to provést pro různých maticových elementů odpovídajících kombinacím různých faktorů časové evoluce. Existují způsoby, jak toto zredukovat, ale v tomto hrubém přiblížení čas potřebný pro tento krok škáluje jako Elementy musí být odhadnuty, což škáluje jako . Nakonec řešení zobecněného problému vlastních čísel v promítnutém prostoru klasicky vyžaduje
Vidíme, že kvantová Krylovská diagonalizace může být užitečná v případech, kdy je počet komutujících Pauliho skupin v Hamiltonianu relativně malý. Tato závislost na škálování naznačuje některé aplikace, kde může být Krylovská metoda užitečná, a jiné, kde pravděpodobně nebude. Některé Hamiloniany mají při zobrazení na qubity vysokou složitost — obsahují mnoho nekomutujících Pauliho řetězců, které nelze snadno rozdělit do několika komutujících skupin. To platí například pro problémy kvantové chemie. Tato složitost přináší dvě hlavní výzvy pro krátkodobé kvantové počítače:
- Odhad každého elementu se stává výpočetně nákladným kvůli velkému počtu členů.
- Požadované Trotterovy obvody se stávají nepřijatelně hlubokými.
Oba výše uvedené body budou méně problematické, až kvantové počítače dosáhnou odolnosti vůči chybám, ale v krátkodobém horizontu je třeba je zohlednit. Stejné překážky mohou postihnout i systémy s „jednodušším" zobrazením než ty v kvantové chemii, pokud Hamiloniany obsahují příliš mnoho nekomutujících členů. Krylovská metoda je nejužitečnější tam, kde lze Hamiltonian rozdělit do relativně mála komutujících Pauliho skupin a kde je snadné implementovat v Trotter obvodech. Obě tyto podmínky jsou splněny například pro mnoho mřížkových modelů, které jsou v fyzice předmětem zájmu. KQD je obzvláště užitečná, když je o základním stavu známo velmi málo. To pramení z jejích inherentních konvergenčních záruk a použitelnosti ve scénářích, kde jsou alternativní metody nepoužitelné kvůli nedostatečné znalosti základního stavu.
Ačkoli je KQD mocným nástrojem, časově náročné aspekty protokolu — zejména odhad každého elementu promítnutého Hamiltonianu a překryvu Krylovských stavů — představují příležitosti ke zlepšení. Alternativní přístup spočívá ve využití Krylovských metod v kombinaci s metodami založenými na vzorkování, které jsou předmětem následující lekce.
6. Přílohy
Appendix I: Krylov subspace from real time-evolutions
Unitární Krylovův prostor je definován jako
pro nějaký časový krok , který určíme později. Předpokládejme dočasně, že je sudé: pak definujme . Všimni si, že při projekci Hamiltoniánu do výše uvedeného Krylovova prostoru ho nelze odlišit od Krylovova prostoru
tedy takového, kde jsou všechny časové evoluce posunuty o časových kroků zpět. Důvodem nerozlišitelnosti je to, že maticové prvky
jsou invariantní vůči celkovým posunům doby evoluce, protože časové evoluce komutují s Hamiltoniánem. Pro liché lze použít analýzu pro .
Chceme ukázat, že někde v tomto Krylovově prostoru se zaručeně nachází stav s nízkou energií. Dokazujeme to pomocí následujícího výsledku, který vychází z Věty 3.1 v [3]:
Tvrzení 1: existuje funkce taková, že pro energie ve spektrálním rozsahu Hamiltoniánu (tedy mezi energií základního stavu a maximální energií)…
- pro všechny hodnoty , které leží od , tedy je exponenciálně potlačena
- je lineární kombinací pro
Důkaz uvádíme níže, ale klidně ho přeskoč, pokud nechceš sledovat celý rigorózní argument. Teď se zaměříme na důsledky výše uvedeného tvrzení. Z vlastnosti 3 výše vidíme, že posunutý Krylovův prostor výše obsahuje stav . To je náš stav s nízkou energií. Abychom pochopili proč, zapišme v energetické vlastní bázi:
kde je k-tý energetický vlastní stav a je jeho amplituda v počátečním stavu . Vyjádřeno pomocí toho, je rovno
s využitím skutečnosti, že lze nahradit , pokud působí na vlastní stav . Energetická chyba tohoto stavu je tedy
Abychom toto převedli na horní mez, která se lépe chápe, nejprve rozdělíme součet v čitateli na členy s a členy s :
První člen můžeme shora omezit hodnotou :
kde první krok plyne z toho, že pro každé v součtu, a druhý krok z toho, že součet v čitateli je podmnožinou součtu ve jmenovateli. Pro druhý člen nejprve odhadneme jmenovatel zdola hodnotou , protože : po sečtení všeho dohromady dostaneme
Abychom zjednodušili zbývající výraz, všimni si, že pro všechna tato víme z definice , že . Dalším odhadem shora a shora dostaneme
To platí pro libovolné , takže pokud nastavíme rovno naší cílové chybě, výše uvedená mez chyby konverguje k ní exponenciálně s Krylovovou dimenzí . Také si všimni, že pokud , pak člen ve výše uvedené mezi ve skutečnosti zcela vymizí.
Pro dokončení argumentu nejprve poznamenejme, že výše uvedené je pouze energetická chyba konkrétního stavu , nikoli energetická chyba stavu s nejnižší energií v Krylovově prostoru. Avšak podle variačního principu (Rayleigh-Ritz) je energetická chyba stavu s nejnižší energií v Krylovově prostoru shora omezena energetickou chybou libovolného stavu v Krylovově prostoru, takže výše uvedené je také horní mezí energetické chyby stavu s nejnižší energií, tedy výstupu algoritmu Krylovovy kvantové diagonalizace.
Podobnou analýzu, jako je ta výše, lze provést tak, aby navíc zohledňovala šum a postup prahování popsaný v notebooku. Viz [2] a [4] pro tuto analýzu.
Appendix II: proof of Claim 1
Následující text je z velké části odvozen z [3], Věta 3.1: nechť a nechť je prostor reziduálních polynomů (polynomů, jejichž hodnota v 0 je 1) stupně nejvýše . Řešením
je
a odpovídající minimální hodnota je
Chceme toto převést na funkci, kterou lze přirozeně vyjádřit pomocí komplexních exponenciál, protože to jsou právě časové evoluce generující kvantový Krylovův prostor. Za tímto účelem je vhodné zavést následující transformaci energií ve spektrálním rozsahu Hamiltoniánu na čísla v rozsahu : definujme
kde je časový krok takový, že . Všimni si, že a roste, jak se vzdaluje od .
Nyní pomocí polynomu s parametry a, b, d nastavenými na , a d = int(r/2) definujeme funkci:
kde je energie základního stavu. Dosazením vidíme, že je trigonometrický polynom stupně , tedy lineární kombinace pro . Z definice výše navíc plyne, že a pro libovolné ve spektrálním rozsahu takové, že , platí
References:
[1] https://arxiv.org/abs/2407.14431
[2] https://arxiv.org/abs/1811.09025
[3] https://people.math.ethz.ch/~mhg/pub/biksm.pdf
[4] https://academic.oup.com/book/36426
[5] https://en.wikipedia.org/wiki/Krylov_subspace
[6] Krylov Subspace Methods: Principles and Analysis, Jörg Liesen, Zdenek Strakos https://academic.oup.com/book/36426
[7] Iterative Methods for Sparse Linear Systems" by Yousef Saad
[8] "MINRES-QLP: A Krylov Subspace Method for Indefinite or Singular Symmetric Systems" by Sou-Cheng Choi, Christopher Paige, and Michael Saunders (https://epubs.siam.org/doi/10.1137/100787921)
[9] Ethan N. Epperly, Lin Lin, and Yuji Nakatsukasa. "A theory of quantum subspace diagonalization". SIAM Journal on Matrix Analysis and Applications 43, 1263–1290 (2022).