Variační kvantový eigensolver (VQE)
Tato lekce představí variační kvantový eigensolver, vysvětlí jeho význam jako základního algoritmu v kvantovém počítání a prozkoumá také jeho silné a slabé stránky. Samotný VQE, bez doplňujících metod, pravděpodobně nebude dostatečný pro moderní kvantové výpočty v měřítku užitečnosti. Přesto je důležitý jako archetypální klasicko-kvantová hybridní metoda a představuje důležitý základ, na kterém je postavena řada pokročilejších algoritmů.
Toto video poskytuje přehled VQE a faktorů, které ovlivňují jeho efektivitu. Text níže přidává další podrobnosti a implementuje VQE pomocí Qiskitu.
1. Co je VQE?
Variační kvantový eigensolver je algoritmus, který k provedení úlohy využívá společně klasické a kvantové počítání. Výpočet VQE má čtyři hlavní součásti:
- Operátor: Často Hamiltonián, který budeme označovat , popisující vlastnost tvého systému, kterou chceš optimalizovat. Jinak řečeno, hledáš vlastní vektor tohoto operátoru, který odpovídá minimální vlastní hodnotě. Tento vlastní vektor často nazýváme „základní stav“.
- „ansatz“ (německé slovo znamenající „přístup“): jde o kvantový obvod, který připravuje kvantový stav aproximující hledaný vlastní vektor. Ve skutečnosti je ansatz rodinou kvantových obvodů, protože některé brány v ansatzu jsou parametrizované, tedy je jim dodáván parametr, který můžeme měnit. Tato rodina kvantových obvodů dokáže připravit rodinu kvantových stavů aproximujících základní stav.
- Estimator: prostředek pro odhad střední hodnoty operátoru nad aktuálním variačním kvantovým stavem. Někdy nám jde skutečně jen o tuto střední hodnotu, které říkáme cenová funkce. Někdy nám jde o složitější funkci, kterou lze přesto zapsat výchozím způsobem z jedné nebo více středních hodnot.
- Klasický optimalizátor: algoritmus, který mění parametry s cílem minimalizovat cenovou funkci.
Podívejme se na každou z těchto součástí podrobněji.
1.1 Operátor (Hamiltonián)
Jádrem úlohy VQE je operátor popisující zkoumaný systém. Budeme zde předpokládat, že nejnižší vlastní hodnota a jí odpovídající vlastní vektor tohoto operátoru jsou užitečné pro nějaký vědecký nebo obchodní účel. Příkladem může být chemický Hamiltonián popisující molekulu, jehož nejnižší vlastní hodnota odpovídá energii základního stavu molekuly a odpovídající vlastní stav popisuje geometrii nebo elektronovou konfiguraci molekuly. Nebo může operátor popisovat náklad určitého procesu, který má být optimalizován, přičemž vlastní stavy mohou odpovídat trasám či postupům. V některých oborech, jako je fyzika, „Hamiltonián“ téměř vždy označuje operátor popisující energii fyzického systému. V kvantovém počítání je však běžné vidět, že jako „Hamiltonián“ jsou označovány i kvantové operátory popisující obchodní nebo logistický problém. Tuto konvenci zde přijmeme.

Mapování fyzikálního nebo optimalizačního problému na qubity je obvykle netriviální úloha, ale tyto podrobnosti nejsou předmětem tohoto kurzu. Obecnou diskusi o mapování problému na kvantový operátor najdeš v Quantum computing in practice. Podrobnější pohled na mapování chemických problémů do kvantových operátorů je k nalezení v Quantum Chemistry with VQE.
Pro účely tohoto kurzu budeme předpokládat, že tvar Hamiltoniánu je známý. Například Hamiltonián pro jednoduchou molekulu vodíku (za jistých předpokladů o aktivním prostoru a s použitím Jordan-Wignerova mapování) je:
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy
from qiskit.quantum_info import SparsePauliOp
hamiltonian = SparsePauliOp(
[
"IIII",
"IIIZ",
"IZII",
"IIZI",
"ZIII",
"IZIZ",
"IIZZ",
"ZIIZ",
"IZZI",
"ZZII",
"ZIZI",
"YYYY",
"XXYY",
"YYXX",
"XXXX",
],
coeffs=[
-0.09820182 + 0.0j,
-0.1740751 + 0.0j,
-0.1740751 + 0.0j,
0.2242933 + 0.0j,
0.2242933 + 0.0j,
0.16891402 + 0.0j,
0.1210099 + 0.0j,
0.16631441 + 0.0j,
0.16631441 + 0.0j,
0.1210099 + 0.0j,
0.17504456 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
],
)
Všimni si, že ve výše uvedeném Hamiltoniánu jsou členy jako ZZII a YYYY, které spolu nekomutují. To znamená, že pro vyhodnocení ZZII bychom potřebovali změřit Pauliho operátor Z na qubitu 3 (mimo jiných měření). Ale pro vyhodnocení YYYY potřebujeme na tom samém qubitu, qubitu 3, změřit Pauliho operátor Y. Mezi operátory Y a Z na témže qubitu existuje relace neurčitosti; nemůžeme oba tyto operátory měřit současně. K tomuto bodu se níže vrátíme, a vlastně i v průběhu celého kurzu.
Výše uvedený Hamiltonián je maticový operátor o rozměrech . Diagonalizace operátoru k nalezení jeho nejnižší energetické vlastní hodnoty není obtížná.
import numpy as np
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues), "hartrees")
The ground state energy is -1.1459778447627311 hartrees
Klasické eigensolvery pracující hrubou silou nedokážou škálovat tak, aby popsaly energie nebo geometrie velmi velkých systémů atomů, jako jsou léky nebo proteiny. VQE je jedním z prvních pokusů, jak v této úloze využít kvantové počítání.
V této lekci se setkáme s Hamiltoniány mnohem většími, než je ten výše. Bylo by však marnotratné posouvat limity toho, co VQE zvládne, dříve než později v tomto kurzu představíme některé pokročilejší nástroje, které mohou VQE doplnit nebo nahradit.
1.2 Ansatz
Slovo „ansatz“ pochází z němčiny a znamená „přístup“. Správný německý plurál je „ansätze“, i když se často setkáme s tvary „ansatzes“ nebo „ansatze“. V kontextu VQE je ansatz kvantový obvod, který používáš k vytvoření mnohaqubitové vlnové funkce co nejvíce aproximující základní stav studovaného systému, a který tak produkuje nejnižší střední hodnotu tvého operátoru. Tento kvantový obvod bude obsahovat variační parametry (často sdružené do vektoru proměnných ).

Zvolí se počáteční množina hodnot variačních parametrů. Unitární operaci ansatzu na obvodu budeme označovat . Ve výchozím nastavení jsou všechny qubity v kvantových počítačích IBM® inicializovány do stavu . Po spuštění obvodu bude stav qubitů
Pokud bychom potřebovali jen nejnižší energii (používáme-li jazyk fyzikálních systémů), mohli bychom ji odhadnout tak, že energii mnohokrát změříme a vezmeme tu nejnižší. Obvykle ale chceme také konfiguraci, která tuto nejnižší energii nebo vlastní hodnotu dává. Dalším krokem je proto odhad střední hodnoty Hamiltoniánu, čehož se dosáhne kvantovými měřeními. To obnáší hodně práce. Kvalitativně ale můžeme tento proces pochopit, pokud si všimneme, že pravděpodobnost naměření energie (opět jazykem fyzikálních systémů) souvisí se střední hodnotou jako:
Pravděpodobnost také souvisí s překryvem mezi vlastním stavem a aktuálním stavem systému :
Tím, že provedeme mnoho měření Pauliho operátorů tvořících náš Hamiltonián, můžeme tedy odhadnout střední hodnotu Hamiltoniánu v aktuálním stavu systému . Dalším krokem je měnit parametry a pokusit se dostat blíže k nejnižšímu energetickému (základnímu) stavu systému. Kvůli variačním parametrům v ansatzu se o něm někdy mluví jako o variační formě.
Než přejdeme k tomuto variačnímu procesu, všimni si, že je často užitečné začít svůj stav z dobře odhadnutého („good guess“) stavu. Možná víš o svém systému dost na to, abys udělal(a) lepší počáteční odhad než . Například v chemických aplikacích je běžné inicializovat qubity do Hartree-Fockova stavu. Tento počáteční odhad, který neobsahuje žádné variační parametry, se nazývá referenční stav. Označme kvantový obvod vytvářející referenční stav jako . Kdykoli je důležité odlišit referenční stav od zbytku ansatzu, použij: Ekvivalentně
1.3 Estimator
Potřebujeme způsob, jak odhadnout střední hodnotu našeho Hamiltoniánu v konkrétním variačním stavu . Kdybychom mohli přímo měřit celý operátor , bylo by to tak jednoduché jako provést mnoho (řekněme ) měření a naměřené hodnoty zprůměrovat:
Symbol nám zde připomíná, že tato střední hodnota by byla přesně správná pouze v limitě . Ale při tisících měření prováděných na obvodu je výběrová chyba střední hodnoty poměrně nízká. Existují další aspekty, například šum, které se stávají problémem u velmi přesných výpočtů.
Obecně však není možné měřit najednou. může obsahovat více nekomutujících Pauliho operátorů X, Y a Z. Hamiltonián se proto musí rozdělit do skupin operátorů, které lze měřit současně, a každá taková skupina se musí odhadovat zvlášť, přičemž výsledky se kombinují za účelem získání střední hodnoty. Tomu se budeme podrobněji věnovat v další lekci, kde budeme diskutovat škálování klasických a kvantových přístupů. Tato složitost měření je jedním z důvodů, proč potřebujeme vysoce efektivní kód pro provádění takového odhadování. V této lekci i dále budeme k tomuto účelu používat primitiv Qiskit Runtime Estimator.
1.4 Klasické optimalizátory
Klasický optimalizátor je jakýkoli klasický algoritmus navržený k nalezení extrémů cílové funkce (obvykle minima). Prohledávají prostor možných parametrů a hledají množinu, která minimalizuje nějakou zajímavou funkci. Lze je zhruba rozdělit na metody založené na gradientu, které využívají informaci o gradientu, a metody bez gradientu, které fungují jako optimalizátory typu černé skříňky. Volba klasického optimalizátoru může významně ovlivnit výkon algoritmu, zvláště v přítomnosti šumu v kvantovém hardwaru. Mezi populární optimalizátory v této oblasti patří Adam, AMSGrad a SPSA, které ukázaly slibné výsledky v šumových prostředích. Mezi tradičnější optimalizátory patří COBYLA a SLSQP.
Běžný pracovní postup (ukázaný v části 3.3) spočívá v použití jednoho z těchto algoritmů jako metody uvnitř minimalizátoru jako je funkce minimize ze scipy. Ta přijímá jako argumenty:
- Nějakou funkci k minimalizaci. Často jde o střední hodnotu energie. Obecně se jim ale říká „cost functions“ (nákladové funkce).
- Množinu parametrů, od které začít hledání. Často označovaná jako nebo .
- Argumenty, včetně argumentů nákladové funkce. V kvantových výpočtech s Qiskitem budou tyto argumenty zahrnovat ansatz, Hamiltonián a primitiv Estimator, o kterém je více řeč v další podkapitole.
- „Metodu“ minimalizace. To odkazuje na konkrétní algoritmus používaný k prohledávání parametrického prostoru. Zde bychom specifikovali například COBYLA nebo SLSQP.
- Volby. Dostupné volby se mohou lišit podle metody. Ale příkladem, který prakticky všechny metody obsahují, je maximální počet iterací optimalizátoru před ukončením hledání: ‚maxiter‘.

V každém iterativním kroku se střední hodnota Hamiltoniánu odhaduje mnoha měřeními. Tuto odhadnutou energii vrací nákladová funkce a minimalizátor aktualizuje informace o energetické krajině. To, co přesně optimalizátor dělá při volbě dalšího kroku, se liší metodu od metody. Některé používají gradienty a vybírají směr nejstrmějšího sestupu. Jiné mohou brát v úvahu šum a mohou vyžadovat, aby se náklady snížily o velkou míru, než přijmou, že skutečná energie podél daného směru klesá.
# Example syntax for minimization
# from scipy.optimize import minimize
# res = minimize(cost_func, x0, args=(ansatz, hamiltonian, estimator), method="cobyla",
# options={'maxiter': 200})
1.5 Variační princip
V tomto kontextu je variační princip velmi důležitý; říká, že žádná variační vlnová funkce nemůže poskytnout střední hodnotu energie (nebo nákladů) nižší, než jakou dává vlnová funkce základního stavu. Matematicky,
Toto lze snadno ověřit, pokud si všimneme, že množina všech vlastních stavů operátoru tvoří úplnou bázi Hilbertova prostoru. Jinými slovy, libovolný stav, a zejména , lze zapsat jako váženou (normalizovanou) sumu těchto vlastních stavů operátoru :
kde jsou konstanty, jež mají být určeny, a . Toto necháváme jako cvičení pro čtenáře. Všimni si ale toho důsledku: variační stav, který produkuje nejnižší střední hodnotu energie, je nejlepším odhadem skutečného základního stavu.
Ověřte si porozumění
Ověř matematicky, že pro libovolný variační stav .
Answer
S využitím daného rozvoje variačního stavu pomocí energetických vlastních stavů,
můžeme variační střední hodnotu energie zapsat jako
Pro všechny koeficienty platí . Můžeme tedy napsat
2. Srovnání s klasickým postupem
Řekněme, že nás zajímá matice s N řádky a N sloupci. Předpokládej, že tvá matice je tak velká, že přesná diagonalizace není možná. Předpokládej dále, že o svém problému víš dost na to, abys mohl/a odhadnout celkovou strukturu cílového vlastního stavu, a chceš prozkoumávat stavy podobné svému počátečnímu odhadu, abys zjistil/a, zda lze cenu/energii dále snížit. Jedná se o variační přístup a je to jedna z metod, která se používá tehdy, když přesná diagonalizace není možná.
2.1 Klasický postup
Na klasickém počítači by to fungovalo takto:
- Vytvoř odhadovaný stav s některými parametry , které budeš měnit: . I když by tento počáteční odhad mohl být náhodný, to se nedoporučuje. Chceme využít znalosti daného problému, abychom náš odhad co nejvíce přizpůsobili.
- Vypočítej střední hodnotu operátoru při stavu systému v tomto stavu:
- Uprav variační parametry a opakuj: .
- Využij nashromážděné informace o prostoru možných stavů ve svém variačním podprostoru k tomu, aby byly odhady stále lepší a lepší a přibližovaly se k cílovému stavu. Variační princip zaručuje, že náš variační stav nemůže vést k vlastní hodnotě nižší, než je hodnota cílového základního stavu. Takže čím nižší je střední hodnota, tím lepší je naše aproximace základního stavu:
Podívejme se na náročnost každého kroku tohoto přístupu. Nastavení nebo aktualizace parametrů je výpočetně snadná; obtíž spočívá ve výběru užitečných, fyzikálně motivovaných počátečních parametrů. Využití nashromážděných informací z předchozích iterací k aktualizaci parametrů tak, abys se přibližoval/a k základnímu stavu, není triviální. Existují ale klasické optimalizační algoritmy, které to dělají poměrně efektivně. Tato klasická optimalizace je nákladná pouze proto, že může vyžadovat mnoho iterací; v nejhorším případě může počet iterací růst exponenciálně s N. Výpočetně nejnákladnějším jednotlivým krokem je téměř jistě výpočet střední hodnoty tvé matice s použitím daného stavu :
Matice musí působit na vektor o N prvcích, což odpovídá: operacím násobení v nejhorším případě. To je třeba provést při každé iteraci parametrů. Pro extrémně velké matice je to výpočetně nákladné.
2.2 Kvantový postup a komutující Pauliovy grupy
Představ si teď, že tuto část výpočtu přenecháš kvantovému počítači. Místo výpočtu této střední hodnoty ji odhaduješ tak, že na kvantovém počítači připravíš stav pomocí svého variačního ansatzu a poté provádíš měření.
To může znít jednodušeji, než to ve skutečnosti je. obecně nelze snadno měřit. Může být například tvořen mnoha nekomutujícími Pauliho operátory X, Y a Z. Ale lze zapsat jako lineární kombinaci členů , z nichž každý je snadno měřitelný (například Pauliho operátory nebo skupiny qubitově komutujících Pauliho operátorů). Střední hodnota v nějakém stavu je váženým součtem středních hodnot jednotlivých členů . Tento výraz platí pro libovolný stav , ale budeme ho konkrétně používat s našimi variačními stavy .
kde je Pauliho řetězec jako IZZX…XIYX, nebo několik takových řetězců, které spolu navzájem komutují. Popis střední hodnoty, který lépe odpovídá realitě měření na kvantových počítačích, je tedy
A v kontextu naší variační vlnové funkce:
Každý z členů lze měřit -krát, čímž získáme vzorky z měření pro , a vrátí střední hodnotu a směrodatnou odchylku . Tyto členy můžeme sečíst a propagovat chyby přes součet, čímž získáme celkovou střední hodnotu a směrodatnou odchylku .
To nevyžaduje žádné rozsáhlé násobení ani žádný proces, který by nutně rostl jako . Místo toho vyžaduje vícenásobná měření na kvantovém počítači. Pokud jich nepotřebuješ příliš mnoho, mohl by být tento přístup efektivní. A to je kvantová část VQE.
Ale pojďme si říct, proč by to nemuselo být efektivní. Jedním z důvodů pro mnoho měření je snížení statistické nejistoty v odhadech u výpočtů s velmi vysokou přesností. Dalším důvodem je počet Pauliho řetězců potřebných k pokrytí celé matice. Protože Pauliho matice (spolu s identitou: X, Y, Z a I) tvoří bázi prostoru všech operátorů dané dimenze, máme zaručeno, že naši matici zájmu lze zapsat jako vážený součet Pauliho operátorů, jak jsme to udělali dříve.
kde je Pauliho řetězec působící na všechny qubity popisující tvůj systém, jako IZZX…XIYX, nebo několik takových řetězců, které spolu navzájem komutují. Připomeň si, že Qiskit používá notaci little endian, ve které -tý Pauliho operátor zprava působí na -tý qubit. Náš operátor tedy můžeme měřit tak, že změříme sérii Pauliho operátorů.
Všechny tyto Pauliho operátory však nelze měřit současně. Pauliho operátory (kromě I) spolu nekomutují, pokud jsou asociovány se stejným qubitem. Například IZIZ a ZZXZ lze měřit současně, protože pro třetí qubit lze měřit I a Z současně, a pro první qubit lze znát I a X současně. Ale ZZZZ a ZZZX nelze měřit současně, protože Z a X nekomutují a oba působí na 0. qubit. Zkušenější čtenáři si možná vzpomenou, že dvě skupiny Pauliho operátorů mohou komutovat jako celek, i když měření jednotlivých qubitů nekomutují. Estimator předpokládá tensorová součinová měření Pauliho operátorů (pomocí rotací báze), což odpovídá seskupování operátorů, které komutují po qubitech. Abychom tedy mohli současně odhadnout dva řetězce (A a B) Pauliho operátorů pomocí Estimatoru, musí Pauliho operátory každého qubitu v A a B komutovat. To znamená, že ZZZZ a ZZXX také nelze měřit současně.

Naši matici tedy rozložíme na součet Pauliho operátorů působících na různé qubity. Některé členy tohoto součtu lze měřit najednou; nazýváme to skupina komutujících Pauliho operátorů. V závislosti na počtu nekomutujících členů může být takových skupin mnoho. Označme počet takových skupin komutujících Pauliho řetězců . Pokud je malé, může to fungovat dobře. Pokud má miliony skupin, nebude to užitečné.
Procesy potřebné pro odhad střední hodnoty jsou shromážděny v primitivu Qiskit Runtime nazvaném Estimator. Více informací o Estimatoru najdeš v referenci API v dokumentaci IBM Quantum®. Estimator lze jednoduše použít přímo, ale Estimator vrací mnohem více než jen nejnižší vlastní hodnotu energie. Vrací například také informace o standardní chybě ansámblu. Proto v kontextu minimalizačních problémů bývá Estimator často součástí nákladové funkce. Více informací o vstupech a výstupech Estimatoru najdeš v tomto průvodci v dokumentaci IBM Quantum.
Zaznamenej střední hodnotu (nebo nákladovou funkci) pro sadu parametrů použitých ve svém stavu a poté parametry aktualizuj. V průběhu času můžeš pomocí odhadnutých středních hodnot nebo hodnot nákladové funkce aproximovat gradient nákladové funkce v podprostoru stavů vzorkovaných tvým ansatzem. Existují jak gradientní, tak bezgradientní klasické optimalizátory. Oba trpí potenciálními problémy s trénovatelností, jako jsou mnohé lokální minima a velké oblasti prostoru parametrů s téměř nulovým gradientem, nazývané barren plateaus.

2.3 Faktory určující výpočetní náklady
VQE nevyřeší všechny tvé nejtěžší problémy kvantové chemie. To ne. Ale cílem není být lepší ve všech výpočtech. Posunuli jsme to, co určuje výpočetní náklady.

Přešli jsme od procesu, jehož složitost závisí pouze na dimenzi matice, k procesu, který závisí na požadované přesnosti a počtu nekomutujících Pauliho operátorů tvořících matici. Poslední část nemá v klasickém výpočetnictví obdobu.
Na základě těchto závislostí může být tento postup užitečný pro řídké matice nebo matice zahrnující málo nekomutujících Pauliho řetězců. To je případ systémů interagujících spinů například. Pro husté matice může být méně užitečný. Víme například, že chemické systémy mají často Hamiltoniány zahrnující stovky, tisíce, dokonce miliony Pauliho řetězců. Bylo provedeno zajímavé práce na snížení tohoto počtu členů. Ale chemické systémy mohou být lépe uzpůsobeny některým z dalších algoritmů, o kterých budeme v tomto kurzu hovořit.
Ověř si porozumění
Uvažuj Hamiltonián na čtyřech qubitech, který obsahuje členy:
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXX
Chceš tyto členy roztřídit do skupin tak, aby všechny členy ve skupině bylo možné měřit současně. Jaký je nejmenší počet takových skupin, které můžeš vytvořit tak, aby byly zahrnuty všechny členy?
Answer
Lze to zvládnout ve 4 skupinách. Všimni si, že taková řešení obvykle nejsou jedinečná.
IIXX, XXXX, IIZZ, ZZZZ
IIXZ, IZXZ, ZIXZ, ZZXZ
IXXZ
XZXZ
Co podle tebe obvykle ztěžuje kvantovou chemii s VQE: počet členů v Hamiltonianu, nebo nalezení dobrého ansatzu?
Answer
Ukazuje se, že existují ansätze, které jsou vysoce optimalizovány pro chemické kontexty. Počet členů v Hamiltonianu, a tedy i počet požadovaných měření, obvykle způsobuje více problémů.
3. Příklad Hamiltoniánu
Pojďme tento algoritmus uvést do praxe na malé hamiltoniánové matici, abychom viděli, co se děje v každém kroku. Použijeme rámec Qiskit patterns:
-Krok 1: Mapování problému na kvantové obvody a operátory -Krok 2: Optimalizace pro cílový hardware -Krok 3: Spuštění na cílovém hardware -Krok 4: Post-procesování výsledků
3.1 Krok 1: Mapování problému na kvantové obvody a operátory
Použijeme Hamiltonián definovaný výše z chemického kontextu. Začneme několika obecnými importy.
# General imports
import numpy as np
# SciPy minimizer routine
from scipy.optimize import minimize
# Plotting functions
import matplotlib.pyplot as plt
Opět předpokládáme, že nás zajímající Hamiltonián je znám. Použijeme zde velmi malý Hamiltonián, protože jiné metody probírané v tomto kurzu budou efektivnější při řešení větších problémů.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
hamiltonian = SparsePauliOp.from_list(
[("YZ", 0.3980), ("ZI", -0.3980), ("ZZ", -0.0113), ("XX", 0.1810)]
)
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues))
The ground state energy is -0.702930394459531
V Qiskitu existuje mnoho předpřipravených voleb ansatz. Použijeme efficient_su2.
# Pre-defined ansatz circuit and operator class for Hamiltonian
from qiskit.circuit.library import efficient_su2
# Note that it is more common to place initial 'h' gates outside the ansatz.
# Here we specifically wanted this layer structure.
ansatz = efficient_su2(
hamiltonian.num_qubits, su2_gates=["h", "rz", "y"], entanglement="circular", reps=1
)
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.decompose().draw("mpl", style="iqp")
This circuit has 4 parameters
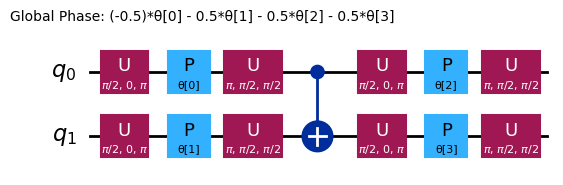
Různé ansätze budou mít různé entanglovací struktury a různá rotační hradla. Zde zobrazený ansatz používá hradla CNOT pro entanglování a hradla Y spolu s parametrizovanými hradly RZ pro rotace. Všimni si velikosti tohoto prostoru parametrů; znamená to, že musíme minimalizovat účelovou funkci přes 4 proměnné (parametry pro hradla RZ). To lze škálovat, ale ne donekonečna. Spuštění podobného problému na 4 qubitech, s použitím výchozích 3 opakování pro efficient_su2, přináší 16 variačních parametrů.
3.2 Krok 2: Optimalizace pro cílový hardware
Ansatz byl napsán s využitím známých hradel, ale náš obvod musí být transpilován, aby využíval bázová hradla, která lze implementovat na každém kvantovém počítači. Vybereme nejméně vytížený backend.
# runtime imports
from qiskit_ibm_runtime import QiskitRuntimeService, Session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# To run on hardware, select the backend with the fewest number of jobs in the queue
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
print(backend)
<IBMBackend('ibm_torino')>
Nyní můžeme transpilovat náš obvod pro tento hardware a vizualizovat náš transpilovaný ansatz.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")

Všimni si, že použitá hradla se změnila a qubity v našem abstraktním obvodu byly namapovány na qubity s jiným číslováním na kvantovém počítači. Abychom dosáhli smysluplných výsledků, musíme stejným způsobem namapovat i náš Hamiltonián.
hamiltonian_isa = hamiltonian.apply_layout(layout=ansatz_isa.layout)
3.3 Krok 3: Spuštění na cílovém hardwaru
3.3.1 Hlášení výstupních hodnot
Zde definujeme účelovou funkci, která přijímá jako argumenty struktury, které jsme sestavili v předchozích krocích: parametry, ansatz a Hamiltonián. Používá také Estimator, který jsme zatím nedefinovali. Zahrnujeme kód pro sledování historie naší účelové funkce, abychom mohli zkontrolovat chování konvergence.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from Estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
Je velmi výhodné, pokud si můžeš zvolit počáteční hodnoty parametrů na základě znalosti daného problému a vlastností cílového stavu. Nepředpokládáme žádné takové znalosti a použijeme náhodné počáteční hodnoty.
x0 = 2 * np.pi * np.random.random(num_params)
# This required 13 min, 20 s QPU time on an Eagle processor, 28 min total time.
with Session(backend=backend) as session:
estimator = Estimator(mode=session)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 50},
)
Iters. done: 1 [Current cost: 0.010575798722044727]
Iters. done: 2 [Current cost: 0.004040015974440895]
Iters. done: 3 [Current cost: 0.0020213258785942503]
Iters. done: 4 [Current cost: 0.18723082446726014]
Iters. done: 5 [Current cost: -0.2746792152068885]
Iters. done: 6 [Current cost: -0.3094547651648519]
Iters. done: 7 [Current cost: -0.05281985428356641]
Iters. done: 8 [Current cost: 0.00808560303514377]
Iters. done: 9 [Current cost: -0.0014821685303514388]
Iters. done: 10 [Current cost: -0.004759824281150161]
Iters. done: 11 [Current cost: 0.09942328705995292]
Iters. done: 12 [Current cost: 0.01092366214057508]
Iters. done: 13 [Current cost: 0.05017497496069776]
Iters. done: 14 [Current cost: 0.13028868414310696]
Iters. done: 15 [Current cost: 0.013747803514376994]
Iters. done: 16 [Current cost: 0.2583072432944498]
Iters. done: 17 [Current cost: -0.14422125655131562]
Iters. done: 18 [Current cost: -0.0004950150347678081]
Iters. done: 19 [Current cost: 0.00681082268370607]
Iters. done: 20 [Current cost: -0.0023377795527156544]
Iters. done: 21 [Current cost: 0.6027665591169237]
Iters. done: 22 [Current cost: 0.00596641373801917]
Iters. done: 23 [Current cost: -0.008318769968051117]
Iters. done: 24 [Current cost: -0.00026683306709265246]
Iters. done: 25 [Current cost: -0.007648222843450479]
Iters. done: 26 [Current cost: 0.004121086261980831]
Iters. done: 27 [Current cost: -0.004075019968051117]
Iters. done: 28 [Current cost: -0.004419369009584665]
Iters. done: 29 [Current cost: 0.213185460054037]
Iters. done: 30 [Current cost: -0.06505919572162797]
Iters. done: 31 [Current cost: -0.5334241316590271]
Iters. done: 32 [Current cost: 0.00218370607028754]
Iters. done: 33 [Current cost: 0.09579352143666908]
Iters. done: 34 [Current cost: -0.009274800319488819]
Iters. done: 35 [Current cost: -0.44395141360688106]
Iters. done: 36 [Current cost: 0.011747104632587858]
Iters. done: 37 [Current cost: -0.003344149361022364]
Iters. done: 38 [Current cost: 0.19138183916486304]
Iters. done: 39 [Current cost: 0.013513931813145209]
Můžeme se podívat na surové výstupy.
res
message: Return from COBYLA because the trust region radius reaches its lower bound.
success: True
status: 0
fun: -0.5334241316590271
x: [ 1.024e+00 6.459e+00 3.625e+00 4.007e+00]
nfev: 39
maxcv: 0.0
3.4 Krok 4: Zpracování výsledků
Pokud postup skončí správně, hodnoty v našem slovníku by měly odpovídat vektoru řešení a celkovému počtu vyhodnocení funkce. To lze snadno ověřit:
cost_history_dict
{'prev_vector': array([1.02397956, 6.45886604, 3.62479262, 4.00744128]),
'iters': 39,
'cost_history': [np.float64(0.010575798722044727),
np.float64(0.004040015974440895),
np.float64(0.0020213258785942503),
np.float64(0.18723082446726014),
np.float64(-0.2746792152068885),
np.float64(-0.3094547651648519),
np.float64(-0.05281985428356641),
np.float64(0.00808560303514377),
np.float64(-0.0014821685303514388),
np.float64(-0.004759824281150161),
np.float64(0.09942328705995292),
np.float64(0.01092366214057508),
np.float64(0.05017497496069776),
np.float64(0.13028868414310696),
np.float64(0.013747803514376994),
np.float64(0.2583072432944498),
np.float64(-0.14422125655131562),
np.float64(-0.0004950150347678081),
np.float64(0.00681082268370607),
np.float64(-0.0023377795527156544),
np.float64(0.6027665591169237),
np.float64(0.00596641373801917),
np.float64(-0.008318769968051117),
np.float64(-0.00026683306709265246),
np.float64(-0.007648222843450479),
np.float64(0.004121086261980831),
np.float64(-0.004075019968051117),
np.float64(-0.004419369009584665),
np.float64(0.213185460054037),
np.float64(-0.06505919572162797),
np.float64(-0.5334241316590271),
np.float64(0.00218370607028754),
np.float64(0.09579352143666908),
np.float64(-0.009274800319488819),
np.float64(-0.44395141360688106),
np.float64(0.011747104632587858),
np.float64(-0.003344149361022364),
np.float64(0.19138183916486304),
np.float64(0.013513931813145209)]}
fig, ax = plt.subplots()
x = np.linspace(0, 10, 50)
# Define the constant function
constant = -0.7029
y_constant = np.full_like(x, constant)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

IBM Quantum nabízí další vzdělávací materiály týkající se VQE. Pokud jsi připraven/a VQE použít v praxi, podívej se na náš tutoriál: Odhadování energie základního stavu Heisenbergova řetězce pomocí VQE. Pokud chceš více informací o vytváření molekulárních Hamiltonánů, podívej se na tuto lekci v našem kurzu Kvantová chemie s VQE. Pokud tě zajímá hlubší pochopení toho, jak variační algoritmy jako VQE fungují, doporučujeme kurz Variational Algorithm Design.
Ověř si znalosti
V této části jsme vypočítali energii základního stavu z Hamiltonianu. Pokud bychom to chtěli použít například k určení geometrie molekuly, jak bychom to rozšířili?
Answer
Museli bychom zavést proměnné pro vzdálenosti mezi atomy a úhly mezi vazbami a tyto proměnné by bylo potřeba měnit. Pro každou jejich variaci bychom vytvořili nový Hamiltonián (protože operátory popisující energii závisejí na geometrii). Pro každý takový Hamiltonián mapovaný na qubity bychom museli provést optimalizaci podobnou té výše. Ze všech těchto mnoha konvergovaných optimalizačních problémů by geometrie s nejnižší energií byla ta, kterou příroda skutečně přijme. To je výrazně složitější než to, co bylo ukázáno výše. Takovýto výpočet je proveden pro nejjednodušší molekulu, , zde.
4. Vztah VQE k jiným metodám
V této části si projdeme výhody a nevýhody původního přístupu VQE a poukážeme na jeho vztahy k jiným, novějším algoritmům.
4.1 Silné a slabé stránky VQE
Některé silné stránky již byly zmíněny. Patří mezi ně:
- Vhodnost pro moderní hardware: Některé kvantové algoritmy vyžadují mnohem nižší chybovost, blížící se rozsáhlé odolnosti vůči chybám. VQE to nevyžaduje; lze ho implementovat na současných kvantových počítačích.
- Mělké obvody: VQE často využívá relativně mělké kvantové obvody. Díky tomu je VQE méně náchylné k akumulovaným chybám hradel a vhodné pro mnoho technik zmírňování chyb. Samozřejmě obvody nejsou vždy mělké; záleží na použitém ansatzu.
- Všestrannost: VQE lze (v zásadě) aplikovat na jakýkoli problém, který lze formulovat jako problém vlastních hodnot/vlastních vektorů. Existuje mnoho upozornění, která VQE pro některé problémy znevýhodňují nebo znemožňují. Některá z nich jsou shrnuta níže.
Některé slabé stránky VQE a problémy, pro které je nepraktické, byly také popsány výše. Patří mezi ně:
- Heuristická povaha: VQE nezaručuje konvergenci ke správné energii základního stavu, protože jeho výkon závisí na volbě ansatz a optimalizačních metod[1-2]. Pokud je zvolen špatný ansatz, kterému chybí potřebné provázání pro požadovaný základní stav, žádný klasický optimalizátor nemůže tento základní stav dosáhnout.
- Potenciálně velký počet parametrů: Velmi expresivní ansatz může mít tolik parametrů, že iterace minimalizace jsou velmi časově náročné.
- Vysoké nároky na měření: Ve VQE se Estimator používá k odhadu střední hodnoty každého členu Hamiltoniánu. Většina Hamiltonánů, o které máme zájem, bude mít členy, které nelze odhadnout současně. To může VQE udělat náročným na zdroje pro velké systémy se složitými Hamiltoniány[1].
- Vliv šumu: Když klasický optimalizátor hledá minimum, hlučné výpočty ho mohou mást a odvádět od skutečného minima nebo zpomalovat jeho konvergenci. Jedním z možných řešení je využití nejmodernějších technik zmírňování a potlačování chyb[2-3] od IBM.
- Barren plateaus: Tyto oblasti mizejících gradientů[2-3] existují i v nepřítomnosti šumu, ale šum je činí problematičtějšími, protože změna středních hodnot způsobená šumem může být větší než změna způsobená aktualizací parametrů v těchto pustých oblastech.
4.2 Vztah k jiným přístupům
Adapt-VQE
Algoritmus ADAPT-VQE (Adaptive Derivative-Assembled Pseudo-Trotter Variational Quantum Eigensolver) je vylepšením původního algoritmu VQE, navržené pro zlepšení efektivity, přesnosti a škálovatelnosti kvantových simulací, zejména v kvantové chemii.
Původní algoritmus VQE popsaný v celé této lekci používá předdefinovaný, pevný ansatz k aproximaci základního stavu systému. V našem případě jsme použili efficient_su2 s jedním opakováním, s rotačními hradly Y a RZ. Ačkoliv se parametry v hradlech RZ měnily, struktura tohoto ansatzu a použitá hradla se neměnily.
ADAPT-VQE řeší omezení VQE prostřednictvím adaptivní konstrukce ansatz. Namísto zahájení s pevným ansatz ADAPT-VQE dynamicky buduje ansatz iterativně. V každém kroku vybere operátor z předdefinovaného souboru (jako jsou operátory fermionové excitace), který má největší gradient vzhledem k energii. To zajišťuje, že jsou přidávány pouze nejdůležitější operátory, což vede ke kompaktnímu a efektivnímu ansatz[4-6]. Tento přístup může mít několik příznivých účinků:
- Snížená hloubka obvodu: Díky postupnému rozrůstání ansatzu a zaměření pouze na nezbytné operátory ADAPT-VQE minimalizuje operace hradel ve srovnání s tradičními přístupy VQE[5,7].
- Zlepšená přesnost: Adaptivní povaha umožňuje ADAPT-VQE obnovit více korelační energie v každém kroku, což ho činí zvláště efektivním pro silně korelované systémy, kde si tradiční VQE nevede dobře[8,9].
- Škálovatelnost a odolnost vůči šumu: Kompaktní ansatz snižuje akumulaci chyb hradel, snižuje výpočetní zátěž a omezuje počet variačních parametrů, které musí být minimalizovány.
ADAPT-VQE stále není dokonalý. V některých případech může uvíznout nebo zpomalit v lokálních minimech a může trpět nadměrnou parametrizací. Může být také poměrně náročný na zdroje, protože vyžaduje výpočet gradientů a optimalizaci parametrů s mnoha strukturami hradel.
Kvantový odhad fáze (QPE)
QPE má podobný účel jako VQE, ale velmi odlišnou implementaci. QPE vyžaduje kvantové počítače odolné vůči chybám kvůli obecně hlubokým kvantovým obvodům a vysoké úrovni koherence, kterou vyžaduje. Jakmile bude možné QPE implementovat, bylo by přesnější než VQE. Jeden způsob popisu rozdílu je prostřednictvím přesnosti jako funkce hloubky obvodu. QPE dosahuje přesnosti s hloubkami obvodu škálovanými jako [10]. VQE vyžaduje vzorků k dosažení stejné přesnosti[10,11].
Krylov, SQD, QSCI a další v tomto kurzu
VQE pomohlo ustanovit kvantové algoritmy, které stále závisejí na klasických počítačích, ne jen pro provoz kvantového počítače, ale pro podstatné části algoritmu. Několik takových algoritmů je středem zájmu zbytku tohoto kurzu. Zde uvádíme letmé vysvětlení několika z nich, jednoduše abychom je porovnali s VQE. Budou podrobněji vysvětleny v následujících lekcích.
Krylovova kvantová diagonalizace (KQD)
Krylovovy metody podprostoru jsou způsoby projekce matice na podprostor za účelem snížení její dimenze a zvýšení její spravovatelnosti při zachování nejdůležitějších vlastností. Jedním trikem v této metodě je generování podprostoru, který tyto vlastnosti zachovává; ukázalo se, že generování tohoto podprostoru úzce souvisí s dobře zavedenou metodou na kvantových počítačích zvanou Trotterizace.
Existuje několik variant kvantových Krylovových metod, ale obecně je přístup následující:
- Použít kvantový počítač ke generování podprostoru (Krylovův podprostor) prostřednictvím Trotterizace
- Promítnout matici zájmu na tento Krylovův podprostor
- Diagonalizovat nový promítnutý Hamiltonián pomocí klasického počítače
Vzorkovací kvantová diagonalizace (SQD)
Vzorkovací kvantová diagonalizace (SQD) souvisí s Krylovovou metodou v tom, že se také pokouší snížit dimenzi matice, která má být diagonalizována, při zachování klíčových vlastností. SQD to provádí následujícím způsobem:
- Začít s počátečním odhadem základního stavu a připravit systém v tomto základním stavu.
- Použít Sampler k vzorkování bitových řetězců, které tvoří tento stav.
- Použít kolekci výpočetních bázových stavů ze vzorkovače jako podprostor, na který promítneš svou matici zájmu.
- Diagonalizovat menší, promítnutou matici pomocí klasického počítače.
To souvisí s VQE v tom, že využívá klasické i kvantové výpočty pro podstatné součásti algoritmu. Oba také sdílejí požadavek na přípravu dobrého počátečního odhadu nebo ansatz. Ale distribuce práce mezi klasickým a kvantovým počítačem v SQD je více podobná Krylovově metodě.
Ve skutečnosti byly Krylovova metoda a SQD nedávno kombinovány do metody vzorkovací Krylovovy kvantové diagonalizace (SKQD) [12].
Kvantová interakce konfiguračního podprostoru
Kvantová vybraná konfigurační interakce (QSCI)[13] je algoritmus, který produkuje aproximovaný základní stav Hamiltoniánu vzorkováním zkušební vlnové funkce za účelem identifikace významných výpočetních bázových stavů pro generování podprostoru pro klasickou diagonalizaci. Jak SQD, tak QSCI používají kvantový počítač ke konstrukci redukovaného podprostoru. Další silnou stránkou QSCI je příprava stavů, zejména v kontextu chemických problémů. Využívá různé strategie, jako je použití časově vyvíjených stavů [14] a souboru chemicky inspirovaných ansätze. Zaměřením na efektivní přípravu stavů QSCI snižuje náklady na kvantové výpočty pro chemické Hamiltoniány při zachování vysoké věrnosti a využití odolnosti vůči šumu ze vzorkovacích technik kvantových stavů [15]. QSCI také poskytuje adaptivní konstrukční techniku, která nabízí více ansätze pro lepší výsledek.
Výchozí pracovní postup QSCI pro chemický problém je následující:
- Sestavit molekulární Hamiltonián pomocí softwaru dle vlastního výběru (například SciPy).
- Připravit algoritmus QSCI výběrem vhodného počátečního stavu a chemicky inspirovaného ansatz s předem vybranou sadou parametrů.
- Vzorkovat významné bázové stavy a diagonalizovat Hamiltonián pomocí klasického počítače k získání energie základního stavu.
- Často se jako technika post-processingu používá obnova konfigurace [16] a symetrická postselekce [15].
- Volitelně má pracovní postup adaptivního QSCI přídavnou optimalizační smyčku od kroku 2 ke kroku 3, pomocí více ansätze s náhodnými počátečními stavy.
Zkontroluj své porozumění
Co mají VQE a všechny ostatní výše uvedené metody společného (kromě QPE, které není podrobně popsáno)
Answer
Všechny zahrnují zkušební stav nebo vlnovou funkci nějakého druhu. Všechny fungují nejlépe, když je počáteční odhad pro tento zkušební stav vynikající.
Další správnou odpovědí je, že všechny jsou nejsnadněji implementovatelné, když je Hamiltonián snadno měřitelný (lze ho rozdělit do relativně mála skupin komutujících operátorů Pauli).
Co nemá VQE společného s žádnou z ostatních výše uvedených metod?
Answer
Klasické optimalizátory. Žádná z ostatních metod nepoužívá algoritmy klasické optimalizace pro výběr variačních parametrů.
Reference
[2] https://en.wikipedia.org/wiki/Variational_quantum_eigensolver
[3] https://journals.aps.org/prapplied/abstract/10.1103/PhysRevApplied.19.024047
[4] https://arxiv.org/abs/2111.05176
[6] https://inquanto.quantinuum.com/tutorials/InQ_tut_fe4n2_2.html
[7] https://www.nature.com/articles/s41467-019-10988-2
[8] https://arxiv.org/abs/2210.15438
[9] https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.6.013254
[10] https://arxiv.org/html/2403.09624v1
[11] https://www.nature.com/articles/s42005-023-01312-y
[13] https://arxiv.org/abs/1802.00171
[14] https://arxiv.org/abs/2103.08505
[15] https://arxiv.org/html/2501.09702v1
[16] https://quri-sdk.qunasys.com/docs/examples/quri-algo-vm/qsci/
[17] https://arxiv.org/abs/2412.13839