Vzorkování-based Krylovova kvantová diagonalizace fermionického mřížkového modelu
Odhad využití: Devět sekund na procesoru Heron r2 (POZNÁMKA: Toto je pouze odhad. Tvůj skutečný čas se může lišit.)
Výsledky učení
Po absolvování tohoto tutoriálu by uživatelé měli rozumět:
- Jak používat SQD Qiskit addon k aproximaci energie základního stavu mřížkového modelu pomocí bitstrings vzorkovaných z kvantového procesoru (QPU).
- Jak používat ffsim k sestavení okruhů časového vývoje pro fermionickou simulaci.
- Jak kombinovat vzorky z více okruhů pro post-processing pomocí algoritmu vzorkování-based Krylovovy diagonalizace (SKQD).
Předpoklady
Doporučujeme, aby si uživatelé před absolvováním tohoto tutoriálu byli obeznámeni s následujícími tématy:
- Vzorkování-based kvantová diagonalizace chemického hamiltoniánu
- Krylovova kvantová diagonalizace mřížkových hamiltoniánů
- Primitiva Qiskit
Pozadí
Tento tutoriál ukazuje, jak použít vzorkování-based kvantovou diagonalizaci (SQD) k odhadnutí energie základního stavu fermionického mřížkového modelu. Konkrétně studujeme jednorozměrný model single-impurity Anderson (SIAM), který se používá k popisu magnetických nečistot zakotvených v kovech.
Tento tutoriál sleduje podobný postup jako příbuzný tutoriál Vzorkování-based kvantová diagonalizace chemického hamiltoniánu. Klíčový rozdíl spočívá v tom, jak jsou sestaveny kvantové Circuit. Druhý tutoriál používá heuristický variační ansatz, který je atraktivní pro chemické hamiltoniány s potenciálně miliony interakčních členů. Na druhou stranu, tento tutoriál používá Circuit, které aproximují časový vývoj podle hamiltoniánu. Takovéto Circuit mohou být hluboké, což tento přístup lépe hodí pro aplikace na mřížkové modely. Stavové vektory připravené těmito Circuit tvoří základ pro Krylovův podprostor, a v důsledku toho algoritmus prokazatelně a efektivně konverguje k základnímu stavu za vhodných předpokladů.
Přístup použitý v tomto tutoriálu lze chápat jako kombinaci technik použitých v SQD a Krylovově kvantové diagonalizaci (KQD). Kombinovaný přístup se někdy označuje jako vzorkování-based Krylovova kvantová diagonalizace (SQKD). Viz Krylovova kvantová diagonalizace mřížkových hamiltoniánů pro tutoriál o metodě KQD.
Tento tutoriál vychází z práce "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization", na kterou se můžeš odkázat pro více podrobností.
Model single-impurity Anderson (SIAM)
Jednorozměrný hamiltonián SIAM je součtem tří členů:
kde
Zde jsou fermionské operátory vytváření/anihilace pro místo lázně se spinem , jsou operátory vytváření/anihilace pro módu nečistoty, a . , a jsou reálná čísla popisující hopping, on-site a hybridizační interakce, a je reálné číslo specifikující chemický potenciál.
Všimni si, že hamiltonián je konkrétní instance obecného hamiltoniánu interagujících elektronů,
kde sestává z jednočásticových členů, které jsou kvadratické ve fermionských operátorech vytváření a anihilace, a sestává z dvočásticových členů, které jsou kvartické. Pro SIAM,
a obsahuje zbytek členů hamiltoniánu. Abychom hamiltonián reprezentovali programově, ukládáme matici a tenzor .
Polohová a impulzová báze
Kvůli přibližné translační symetrii v neočekáváme, že základní stav bude řídký v polohové bázi (orbitální báze, v níž je hamiltonián výše specifikován). Výkon SQD je zaručen pouze tehdy, pokud je základní stav řídký, tj. má výraznou váhu pouze na malém počtu stavů výpočetní báze. Abychom zlepšili řídkost základního stavu, provádíme simulaci v orbitální bázi, ve které je diagonální. Tuto bázi nazýváme impulzová báze. Protože je kvadratický fermionský hamiltonián, lze jej efektivně diagonalizovat orbitální rotací.
Přibližný časový vývoj podle hamiltoniánu
K aproximaci časového vývoje podle hamiltoniánu používáme Trotter-Suzukiho rozklad druhého řádu,
Pomocí Jordanovy-Wignerovy transformace se časový vývoj podle rovná jednomu Gate CPhase mezi spin-up a spin-down orbitaly na místě nečistoty. Protože je kvadratický fermionský hamiltonián, časový vývoj podle odpovídá orbitální rotaci.
Krylovovy bázové stavy , kde je dimenze Krylovova podprostoru, jsou tvořeny opakovanou aplikací jednoho Trotterova kroku, takže
V následujícím pracovním postupu založeném na SQD budeme vzorkovat z této sady Circuit a post-procesovat kombinovanou sadu bitstrings pomocí SQD. Tento přístup je v kontrastu s přístupem používaným v příbuzném tutoriálu Vzorkování-based kvantová diagonalizace chemického hamiltoniánu, kde byly vzorky odebírány z jednoho heuristického variačního Circuit.
Požadavky
Před zahájením tohoto tutoriálu se ujisti, že máš nainstalováno následující:
- Qiskit SDK v1.0 nebo novější, s podporou vizualizace
- Qiskit Runtime v0.22 nebo novější (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 nebo novější (
pip install qiskit-addon-sqd) - ffsim v0.0.72 nebo novější (
pip install ffsim)
Příklad s malým simulátorem
Krok 1: Namapuj problém na kvantový Circuit
Nejprve vygenerujeme hamiltonián SIAM v polohové bázi. Hamiltonián je reprezentován maticí a tenzorem . Poté jej rotujeme do impulzové báze. V polohové bázi umístíme nečistotu na první místo. Když však rotujeme do impulzové báze, přesuneme nečistotu na centrální místo, abychom usnadnili interakce s ostatními orbitaly.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
Dále vygenerujeme Circuit pro přípravu Krylovových bázových stavů. Pro každý spinový druh je počáteční stav dán superpozicí všech možných excitací tří elektronů nejbližších Fermiho hladině do 4 nejbližších prázdných módů, vycházejících ze stavu , realizovaných aplikací sedmi XXPlusYYGate. Časově vyvinuté stavy jsou produkovány postupnými aplikacemi Trotterova kroku druhého řádu.
Pro podrobnější popis tohoto modelu a způsobu, jakým jsou Circuit navrženy, viz "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization".
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
Krok 2: Optimalizace problému pro kvantové spuštění
Dále optimalizujeme Circuit pro cílový hardware. Prozatím vytvoříme generický backend s určeným počtem qubitů a sadou Gate, do které se Circuit časového vývoje přirozeně rozkládají.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
Krok 3: Spuštění pomocí primitiv Qiskit
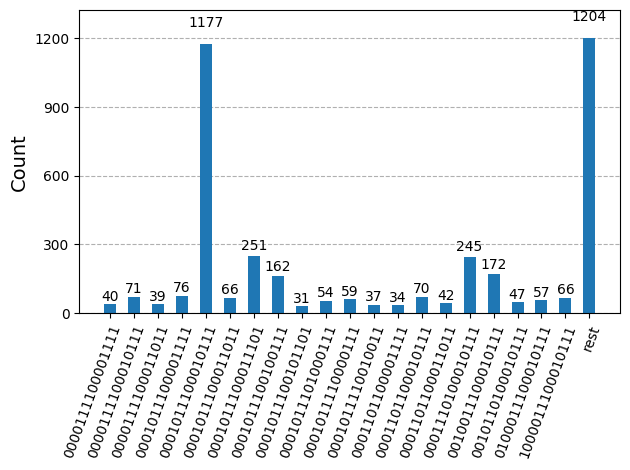
Po optimalizaci okruhů pro spuštění na hardware jsme připraveni spustit je na cílovém hardware a shromáždit vzorky pro odhad energie základního stavu. Po použití primitivy Sampler k vzorkování bitových řetězců z každého okruhu zkombinujeme všechny výsledky do jediného slovníku počtů a vykreslíme 20 nejčastěji vzorkovaných bitových řetězců.
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
Krok 4: Následné zpracování a vrácení výsledku do požadovaného klasického formátu
Teď spustíme algoritmus SQD pomocí funkce diagonalize_fermionic_hamiltonian. Vysvětlení argumentů této funkce najdeš v dokumentaci API.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
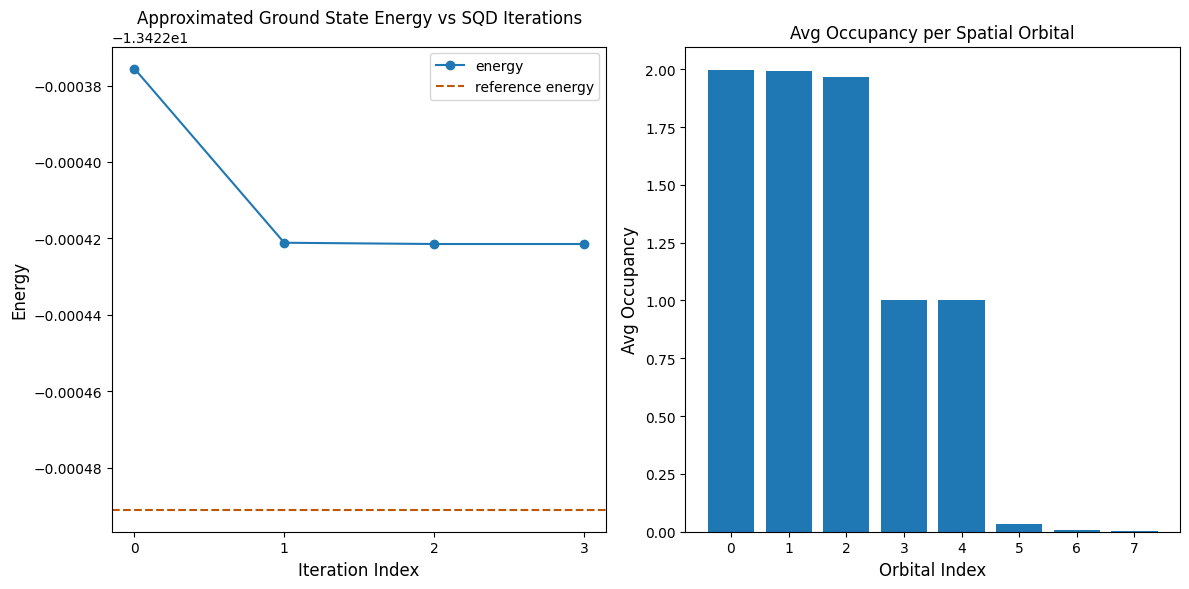
Následující buňka kódu vykresluje výsledky. První graf zobrazuje vypočtenou energii jako funkci počtu iterací obnovy konfigurace, druhý graf zobrazuje průměrnou obsazenost každého prostorového orbitalu po poslední iteraci. Protože je to takto malý problém, první iterace nás již přivede velmi blízko přesné energii (všimni si škály osy y).
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
Ověření energie
Energie vrácená algoritmem SQD je zaručeně horní mezí skutečné energie základního stavu. Hodnotu energie lze ověřit, protože SQD také vrací koeficienty vektoru stavu aproximujícího základní stav. Energii z vektoru stavu můžeš vypočítat pomocí jeho jednočásticových a dvoučásticových redukovaných hustotních matic, jak ukazuje následující buňka kódu.
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
Příklad ve velkém měřítku na hardware
Nyní spustíme větší příklad na reálném QPU. Jako referenční energii používáme výsledky výpočtu metodou DMRG, který byl proveden samostatně.
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
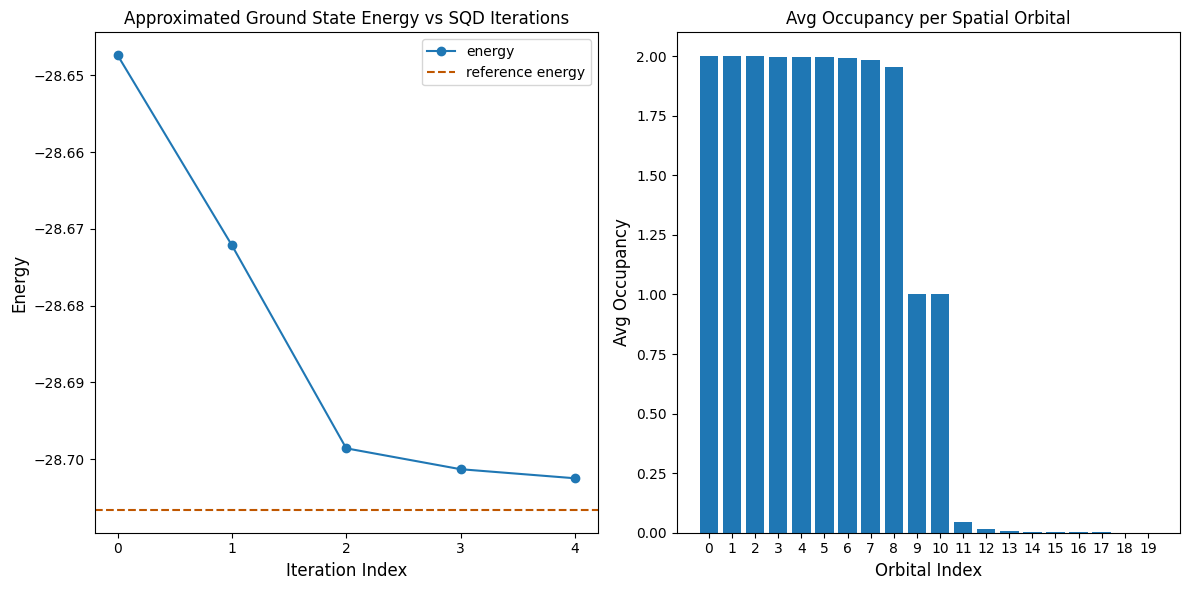
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
Další kroky
Pokud tě tato práce zaujala, mohlo by tě zajímat následující materiál:
- Vzorkování-based kvantová diagonalizace chemického hamiltoniánu – příbuzný tutoriál používající heuristický variační ansatz místo Trotterových okruhů
- Krylovova kvantová diagonalizace mřížkových hamiltoniánů – tutoriál o metodě KQD
- Dokumentace API addonu SQD – reference pro funkci
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization – článek, na němž je tento tutoriál založen