Kvantová diagonalizace na základě vzorků pro chemický Hamiltonián
Odhadovaná spotřeba: méně než jedna minuta na procesoru Heron r2 (POZNÁMKA: Jde pouze o odhad. Skutečná doba běhu se může lišit.)
Výsledky učení
Po absolvování tohoto tutoriálu by uživatelé měli rozumět:
- Jak použít SQD Qiskit addon k aproximaci energie základního stavu molekulárního systému pomocí bitových řetězců vzorkovaných z kvantové procesorové jednotky (QPU).
- Jak použít ffsim k sestavení Circuit lokálního unitárního shlukového Jastrow (LUCJ) pro simulaci kvantové chemie.
Předpoklady
Doporučujeme, aby se uživatelé před absolvováním tohoto tutoriálu seznámili s následujícími tématy:
- Kvantová chemie a druhá kvantizace
- Použití primitiva Sampler k vzorkování z kvantových obvodů
Pozadí
V tomto tutoriálu ukážeme, jak post-procesovat hlučné kvantové vzorky k aproximaci základního stavu molekuly dusíku při rovnovážné délce vazby pomocí SQD Qiskit addonů k implementaci algoritmu kvantové diagonalizace na základě vzorků (SQD). Více podrobností o softwaru najdeš v příslušné dokumentaci, včetně jednoduchého příkladu pro začátek.
Tento tutoriál je doporučen uživatelům, kteří jsou obeznámeni s kvantovou chemií: konkrétně s hledáním energií základního stavu molekuly. Podrobný průvodce pracovním postupem najdeš v kurzu algoritmu kvantové diagonalizace.
SQD je technika pro hledání vlastních hodnot a vlastních vektorů kvantových operátorů, jako je Hamiltonián kvantového systému, pomocí kombinace kvantového a distribuovaného klasického výpočtu. Klasický distribuovaný výpočet slouží ke zpracování vzorků získaných z kvantového procesoru a k projekci a diagonalizaci cílového Hamiltoniánu v podprostoru jimi generovaném. Pracovní postup SQD se skládá z následujících kroků:
- Zvol Circuit ansatz a aplikuj ho na kvantovém počítači na referenční stav (v tomto případě stav Hartree-Fock).
- Vzorkuj bitové řetězce z výsledného kvantového stavu.
- Spusť proceduru self-consistent configuration recovery na bitových řetězcích, aby ses dostal k aproximaci základního stavu.
Je známo, že SQD funguje dobře, když je cílový vlastní stav řídký: vlnová funkce je podpořena na množině bázových stavů , jejíž velikost neroste exponenciálně s velikostí problému.
Kvantová chemie
Hamiltonián molekulárního systému lze zapsat jako
kde a jsou komplexní čísla nazývaná molekulární integrály, která lze vypočítat ze specifikace molekuly pomocí počítačového programu. V tomto tutoriálu počítáme integrály pomocí softwarového balíku PySCF.
Podrobnosti o tom, jak se odvozuje molekulární Hamiltonián, najdeš v učebnici kvantové chemie (například Modern Quantum Chemistry od Szaba a Ostlunda). Přehledné vysvětlení, jak se problémy kvantové chemie mapují na kvantové počítače, nabízí přednáška Mapping Problems to qubits z Qiskit Global Summer School 2024.
Ansatz lokálního unitárního shlukového Jastrow (LUCJ)
SQD vyžaduje ansatz kvantového obvodu, z něhož se vzorkuje. V tomto tutoriálu použijeme ansatz lokálního unitárního shlukového Jastrow (LUCJ) díky jeho kombinaci fyzikální motivace a přátelskosti k hardwaru. K sestavení Circuit ansatzu použijeme ffsim.
Ansatz LUCJ se přizpůsobuje QPU s omezeným propojením Qubitů. Spinové orbitaly jsou mapovány na Qubity tak, aby ansatz nevyžadoval routing pomocí SWAP Gate. Hardware IBM® má topologii Qubitů heavy-hex mřížky, v tomto případě můžeme použít vzor „cik-cak", znázorněný níže. V tomto vzoru jsou orbitaly stejného spinu mapovány na Qubity s topologií přímky (červené a modré kroužky) a spojení mezi orbitaly různého spinu se vyskytuje každé 4. prostorové orbitaly, přičemž spojení je zprostředkováno ancilárním Qubitem (fialové kroužky).
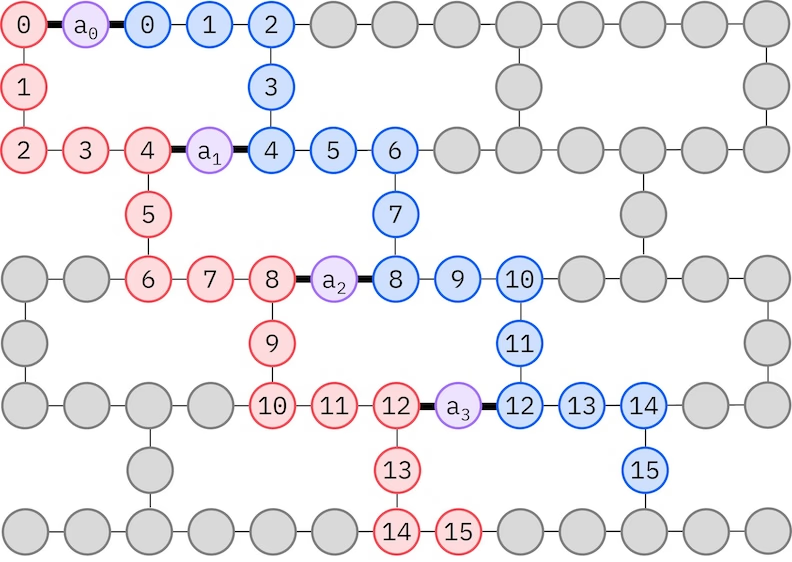
Self-consistent configuration recovery
Procedura self-consistent configuration recovery je navržena k extrakci co největšího signálu z hlučných kvantových vzorků. Protože molekulární Hamiltonián zachovává počet částic a spin Z, dává smysl volit Circuit ansatz, který tyto symetrie také zachovává. Při aplikaci na stav Hartree-Fock má výsledný stav v bezhlučném nastavení pevný počet částic a spin Z. Proto by spin- a spin- části každého bitového řetězce vzorkovaného z tohoto stavu měly mít stejnou Hammingovu váhu jako ve stavu Hartree-Fock. Kvůli přítomnosti šumu v současných kvantových procesorech budou některé naměřené bitové řetězce tuto vlastnost porušovat. Jednoduchá forma postselektion by tyto bitové řetězce zahodila, ale to je plýtvání, protože mohou stále obsahovat určitý signál. Procedura self-consistent recovery se pokouší část tohoto signálu získat zpět v post-processingu. Procedura je iterativní a jako vstup vyžaduje odhad průměrného obsazení každého orbitalu v základním stavu, který se nejprve vypočítá ze surových vzorků. Procedura běží ve smyčce a každá iterace má tyto kroky:
- Pro každý bitový řetězec, který porušuje zadané symetrie, překlop jeho bity pravděpodobnostní procedurou navrženou tak, aby bitový řetězec přiblížila k aktuálnímu odhadu průměrného obsazení orbitalů, a získej nový bitový řetězec.
- Shromáždi všechny staré a nové bitové řetězce, které splňují symetrie, a vzorkuj podmnožiny pevné velikosti, zvolené předem.
- Pro každou podmnožinu bitových řetězců promítni Hamiltonián do podprostoru generovaného odpovídajícími bázovými vektory (popis těchto bázových vektorů viz předchozí sekci) a vypočítej odhad základního stavu promítnutého Hamiltoniánu na klasickém počítači.
- Aktualizuj odhad průměrného obsazení orbitalů pomocí odhadu základního stavu s nejnižší energií.
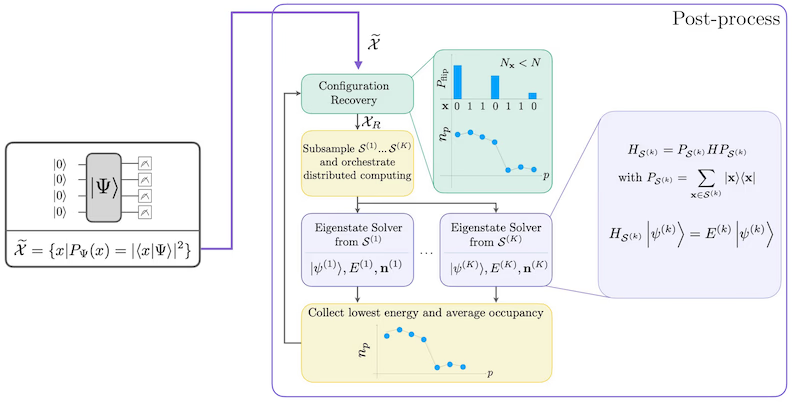
Diagram pracovního postupu SQD
Pracovní postup SQD je znázorněn v následujícím diagramu:
Požadavky
Před zahájením tohoto tutoriálu se ujisti, že máš nainstalováno následující:
- Qiskit SDK v1.0 nebo novější, s podporou vizualizace
- Qiskit Runtime v0.22 nebo novější (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 nebo novější (
pip install qiskit-addon-sqd) - ffsim v0.0.75 nebo novější (
pip install ffsim)
Nastavení
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Příklad na malém simulátoru
V tomto tutoriálu najdeme aproximaci základního stavu molekuly dusíku v blízkosti její rovnovážné délky vazby. Nejprve použijeme malou bázi STO-6G, abychom mohli simulovat experiment a ověřit jeho správnost.
Krok 1: Mapování klasických vstupů na kvantový problém
Nejprve specifikujeme molekulu a její vlastnosti.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Před sestavením Circuit ansatzu LUCJ nejprve provedeme výpočet CCSD v následující buňce kódu. Amplitudy a z tohoto výpočtu budou použity k inicializaci parametrů ansatzu.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Nyní použijeme ffsim k vytvoření Circuit ansatzu. Protože naše molekula má uzavřený stav Hartree-Fock, použijeme spin-vyvážený variant ansatzu UCJ, UCJOpSpinBalanced. V metodě from_t_amplitudes nastavíme optimize=True, aby bylo povoleno „komprimované" dvojité faktorizování amplitud (viz The local unitary cluster Jastrow (LUCJ) ansatz v dokumentaci ffsim pro podrobnosti).
Protože se ansatz LUCJ přizpůsobuje dostupnému propojení QPU, musíme před vytvořením ansatzu inicializovat backend QPU. Prozatím vytvoříme generický backend s coupling mapou heavy-hex a sadou Gate, do které se ansatz LUCJ přirozeně rozkládá. Poté použijeme ffsim.qiskit.generate_lucj_pass_manager k vytvoření pass manageru specializovaného pro transpilaci ansatzu LUCJ na daný backend podle rozložení „cik-cak" popsaného v sekci pozadí o ansatzu LUCJ. Tato funkce používá heuristiku skóre k minimalizaci chyb spojených s vybraným rozložením, což je důležité, pokud je tvůj backend skutečnou QPU nebo simulátorem s modelem šumu. Kromě vrácení pass manageru tato funkce vrací také páry vazeb alpha-beta, které lze na hardwaru implementovat. Pokud nelze implementovat všechny páry, vydá varování.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Krok 2: Optimalizace pro spuštění na kvantovém hardwaru
Dále optimalizujeme Circuit pro cílový hardware. Obvykle tento krok zahrnuje inicializaci hardwarového backendu a pass manageru pro daný backend. Protože se však ansatz LUCJ přizpůsobuje propojení hardwaru, tyto akce jsme již provedli v předchozím kroku. Zbývá pouze spustit pass manager na obvod a transpilovat ho na ISA Circuit, který lze přímo spustit na QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Krok 3: Spuštění pomocí Qiskit primitiv
Po optimalizaci Circuit pro spuštění na hardwaru jsme připraveni spustit ho na cílovém hardwaru a shromáždit vzorky pro odhad energie základního stavu. Protože máme pouze jeden obvod, použijeme režim spuštění úlohy Qiskit Runtime a spustíme náš obvod.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Krok 4: Post-processing a vrácení výsledku v požadovaném klasickém formátu
Užitečnou metrikou pro posouzení kvality výstupu QPU je počet vrácených platných konfigurací. Platná konfigurace má správný počet částic a spin Z, což znamená, že pravá polovina bitového řetězce má Hammingovu váhu rovnou počtu elektronů se spinem nahoru a levá polovina má Hammingovu váhu rovnou počtu elektronů se spinem dolů. Následující buňka vypočítá podíl vzorkovaných konfigurací, které jsou platné.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Všechny bitové řetězce jsou platné, protože vzorkujeme Circuit na bezhlučném simulátoru. Při spuštění na hlučné QPU bude tento podíl nižší než jedna, ale doufejme, že bude vyšší než podíl, který bychom očekávali, kdyby byly bitové řetězce vzorkovány rovnoměrně náhodně, což je vypočítáno v následující buňce.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Nyní odhadneme energii základního stavu Hamiltoniánu pomocí funkce diagonalize_fermionic_hamiltonian. Tato funkce provádí postup self-konzistentního obnovení konfigurací, který iterativně zpřesňuje zašuměné kvantové vzorky za účelem zlepšení odhadu energie. Předáváme callback funkci, abychom mohli uložit mezivýsledky pro pozdější analýzu. Vysvětlení argumentů funkce diagonalize_fermionic_hamiltonian najdeš v dokumentaci API.
Zde použijeme argument initial_occupancies funkce diagonalize_fermionic_hamiltonian k zadání konfigurace Hartree-Fock jako počátečního odhadu obsazení orbitalů v základním stavu. Tento přístup je vhodný pro systémy, kde má základní stav významnou podporu na konfiguraci Hartree-Fock, ale nemusí být vhodný v jiných situacích, ačkoli pokročilejší výpočetní metody mohou v takových případech poskytnout lepší počáteční odhady. Zadání initial_occupancies také umožňuje spuštění obnovy konfigurace, i když nebyly vzorkovány žádné platné konfigurace, což může nastat při vzorkování velkého Circuit na hlučné QPU. Bez tohoto argumentu by obnova konfigurace selhala a vyvolala chybu, pokud by nebyly poskytnuty žádné platné konfigurace.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
Vizualizace výsledků
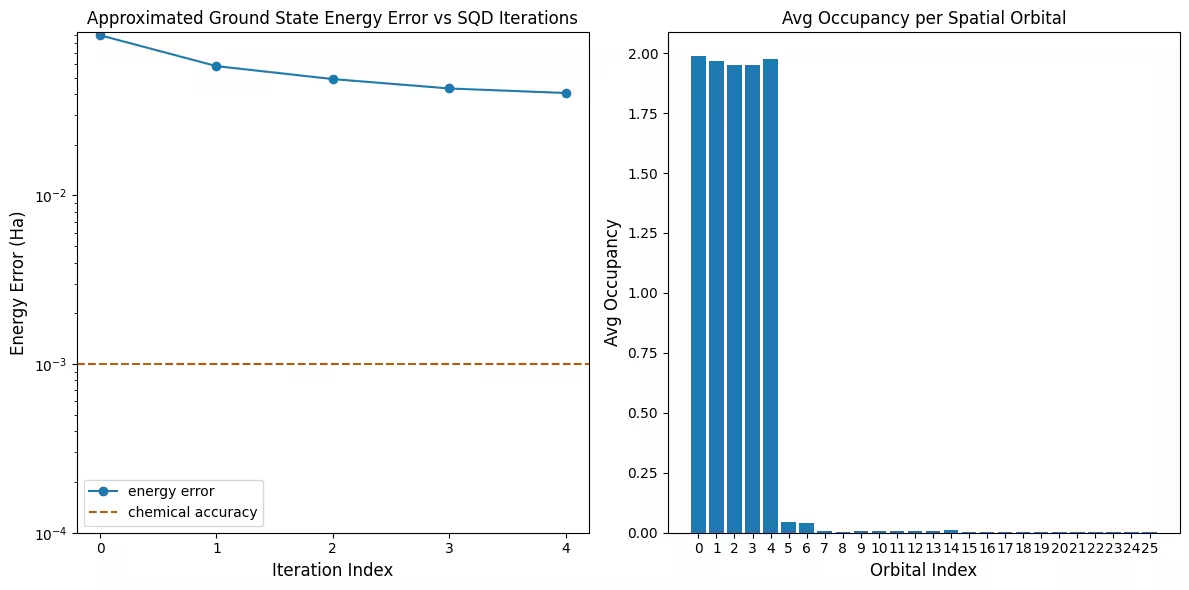
První graf ukazuje, že v této simulaci jsme již po první iteraci v rámci 1 mH od přesné odpovědi (chemická přesnost je typicky považována za 1 kcal/mol 1,6 mH). Jedná se však o malý systém a protože vzorky jsou bezhlučné, obnova konfigurace není potřeba. U většího systému spuštěného na hlučné QPU může být potřeba více iterací obnovy konfigurace a výsledná přesnost může být horší. Obecně platí, že energii lze zlepšit povolením více iterací obnovy konfigurace nebo zvýšením počtu vzorků na dávku.
Druhý graf zobrazuje průměrnou obsazenost každého prostorového orbitalu po poslední iteraci. Vidíme, že elektrony se spinem nahoru i se spinem dolů obsazují s vysokou pravděpodobností prvních pět orbitalů v našich řešeních.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Příklad na velkém hardwaru
Nyní spustíme větší příklad na skutečném kvantovém hardwaru. Zde odvodíme aktivní prostor pro molekulu dusíku z báze cc-pVDZ.
Kroky 1–4
Zde shrneme všechny kroky do jednoho pracovního postupu ve větším měřítku, který je poté spuštěn na skutečném kvantovém hardwaru.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Další kroky
Pokud tě tato práce zaujala, možná tě budou zajímat i následující materiály:
- Kvantová diagonalizace na základě vzorků metodou Krylov pro fermionový mřížkový model – příbuzný tutoriál využívající Circuit s časovým vývojem namísto variačního ansatzu
- Škálování pracovních postupů SQD pro chemii pomocí solveru Dice – stránka ukazující, jak používat efektivnější software Dice pro diagonalizaci
- Dokumentace API SQD addonů – reference pro funkci
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer – článek, na němž je tento tutoriál založen